



TOPICS
CHNフォーラム
第十三部 超微量分析の技法
1.はじめに
20世紀の始めにプレーグル (F. Pregl, 1869~1930) が微量分析法を築き上げてから,その技術は広く世界に伝わり,有機物,特に天然物化学と生化学の進歩に大きな貢献をしてきました。プレーグル自身は医学者で,胆石の研究をしていましたが,たまたま構成成分である胆汁酸の一種と思われる微量の結晶を取り出しました。当時は化学構造の決定に必要な分析試料は一回あたり数グラムであったので,研究を進めるには大量の胆石を集めるか,それとも研究を放棄するかの岐路に立たされました。プレーグルはその時,微量の試料で分析が出来る技術を開発すれば当面の問題だけでなく,同じ問題を抱える天然物,生化学物質の研究に広く活用できると考え,医学者の立場を捨てて微量分析法の研究に生涯没頭しました。プレーグルは一連の微量分析技術を纏めて一冊のテキストとして出版しましたが1),その内容を知って大勢の研究者がさらに改良を試みました。しかしこれらは皆基本的にはマニュアル分析で,部分的な進歩はありましたが,こうして20世紀の前半が推移しました.プレーグルの微量分析法はかなり複雑な操作と洗練された技を必要とするので,この特殊技術を身につけた人々は,余人の真似できない達人とされ,それなりの社会的評価を受けました。
1960年代に始まった機器分析の流れは,電子部品の進歩に支えられて急速に発展し,電気分析,光分析,クロマト分析の分野で毎年のように新しい器械が生まれました。有機元素分析の機器化もこれに刺激されて研究が進み,まずクロマト分離方式のCH分析装置が1960年発表され,数年を経ずしてCHNの同時分析装置に発展しています。測定方式もクロマトに捉われず,赤外スペクトル法や吸着法,さらには差動熱伝導度法も取り入れられて現在に至っています。どの分析機器でも共通してセンサーが搭載され,化学情報を微弱な電気信号に変え,増幅,記録するレコーダーが付属する構成が定着しました。機器の性能は当然原理的なものに支配されますが,同時に電子機器としての信頼性や安定性,高感度と低雑音,データ処理機能など従来の化学分析には無かった要素が加わりました2)。
振り返ると20世紀の前半は分析法の微量化と普及で歴史的な足跡を残し,後半は分析装置の電子化でマニュアル分析からコンピュータ制御の自動分析へと大きく変貌したと言えます。さてそれでは21世紀の現在,次に何を目指すべきかについてそろそろ考えなければなりません。一例を挙げると,分析対象の化学種がC,H,Nのほかハロゲン,硫黄など限られたものになっていますが,周期律表の百何十の元素名を見ているといかにも偏っていて,もっと対象を広げるべきであると考えられます。近年へテロ元素を含む有機化合物が新しい機能を求めて多数合成されていますが,ヘテロ元素自身の定量や,ヘテロ元素を含む有機化合物のCHN分析がうまく出来るかどうかなどの立証が十分ではありません。
他には分析法のスケールアップとスケールダウンの問題があります。スケールアップについては,食品,飼料,工業材料などの品質管理に10~100gの試料を燃焼できるマクロ分析が求められています。またスケールダウンについては天然物化学,生化学部門で0.5 mg以下の試料による超微量分析の手法が要望されています。また視点を変えると,高度の機器分析が手近に利用できるようになっていますので,これらと合体して新しい分析システムを創造することも夢ではないでしょう。一足先にクロマトと質量分析が合体してGC-MSやLC-MSなどが華やかな展開を見せています。地味な行き方として,日常化学操作の中で思いがけないヒントが見つかったりすることもあり,例えば三角フラスコを使った酸素フラスコ燃焼法の貢献は実に素晴らしいものがありました。次世代を支える人々にこれからの事をお願いしたいところですが,本稿では筆者が昔試みたスケールダウン,すなわち分析法の超微量化と周辺情報を取り上げ,表舞台に出なかった技術を参考のため記述したいと思います。
2.超微量試料の計量
プレーグルの微量分析はクールマン(W. Kuhlmann)の微量はかりを手に入れたことから始まりました.当時プレーグルのいたオーストリアのグラーツ大学の近くにグラーツ工科大学があり,そこでエミッヒ(F. Emich, 1860~1940)が化学操作の微量化を研究していました。この研究室ではドイツのハンブルグで作られたクールマンの精密はかりが使われていましたが,大体今日で言うセミマイクロはかりで,プレーグルの目指す3~5 mgの試料の計量には精度の点で満足できませんでした。最大荷重20 g,読み取り精度1μgの高度の要求をプレーグルから受けて間もなくクールマンは歴史的な「クールマン微量はかり」を完成させました1)。このはかりはその後世界中に普及し,わが国にも昭和期の初め多くの大学,研究所に納入されました2)。ドイツ職人技の極致とも言うべきものですが,それだけに測定操作は厳密な手順に従うもので,大雑把な性格の人はとても運用が出来ないものでした。このはかりがうまく動かないと微量分析が出来ないので,ある大学の分析室では毎年正月に鏡餅をはかりの傍に供えてその年の無事な動作を祈っていました。
クールマン微量はかりは棹(さお)が支点を中心に回転振動するもので,棹に取り付けた指針の回帰点を数回読み取り,平均値を静止点とするものですから,ひどく目が疲れ,分析者の苦痛の種でしたが,間もなく空気ダンパーを備えたはかりが市販されるようになり,測定は大分楽になりました。光学拡大による投影像で読み取る方式も加わり,特色ある機種がいろいろ市販されるようなって選択の幅も広がりました2)。こうして微量はかりは短期間に迅速,正確,便利な測定機構を獲得し,その後の微量分析の発展に大きく寄与しました。
微量はかりの進歩と平行して,1940年代にはもっと精密なはかりの研究が始まりました。しかし刃先支持の機構を採用する限り,刃と刃受けの接触面での摩擦が利いて棹の傾きの再現性に限界があり,このため細い石英線ではかりの棹を吊るし,荷重によって傾いた分を石英線の捻りで復元するトーション方式が多く検討されました3, 4)。石英線の径を細くすれば理論上いくらでも感度を上げることが出来ますが,荷重が大きいと切れてしまうので限度があります。石英トーションはかりは間もなく実用品が市販されるようになり,筆者も1961年スエーデン留学中その一台を毎日操作する機会を得ました。アメリカのマイクロテック社製Rodder Ultramicrobalance Model E (Rodderは製作者の名前)という機種で,規格値は最大荷重200 mg,読み取り限度0.05μgでした。帰国して同じものを1台購入し,その後の超微量分析の研究に随分役に立ちました。
この超微量はかりは径0.2~0.3 mmの石英棒を溶接して組み立てた棹が厚いアルミニウムの箱に格納してあり,棹の重心位置付近で径20~30μmの石英線を用い,棹と直角方向に水平に引っ張って吊るしています(図1)。
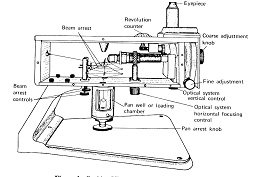
図1.Rodder Model E
棹の両端からは石英製の皿つりが箱の底から垂下していて,開閉窓から試料を皿に載せたり下ろしたり出来ます.左右の荷重の違いによる棹の傾きは石英線の回転で復元しますが,復元はアイピースを覗いて二本の標線を一致 させることで判定できます。回転角はバーニア目盛りを使って精密に読み取りますが,予め標準分銅を使って1目盛り当たりの質量を決めておきます。このはかりの標準偏差は0.06~0.09μgで,空掛けから最大荷重の200 mgまであまり変わりませんでした4)。100~500μgの試料を用いる超微量分析には十分な計量精度があります。
溶融石英を引いて作られた石英線は,捻り角と回転トルクの比例性が極めて良好で,しかも膨張係数がゼロに近いことから,温度による感度変化が殆どありません。購入時標準分銅を使って決めた感度係数は,十数年経過してもう一度感度係数を求めても全く同じであったので,その動作特性の持続性が優れていることに感銘を受けた記憶があります。Rodder Model Eは製作が極めて困難で,20数台が販売されただけで終わりましたが,代わりに接合部をセメントで固めたイギリス製のOertling Model Q 01が生産されるようになって,わが国でもかなり使われました(図2)。接合部が溶接でないので,外れる事故があり,取り扱いには注意が必要です。

図2 Oertling Model Q01
1957年トーションはかりの復元を捻りでなく,電気力で行う電子はかりが開発されました5)。棹の支持部にコイルを取り付け,磁石の間隙に入れて電流を通じると回転力を生じます.荷重による棹の傾きを復元するに要する電流量から質量が分かります(図3)。
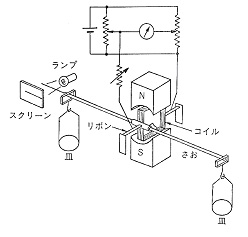
図3.初期の微量電子はかり
棹の支持体はリボン状で,電流を通じるため導電性と弾力性が要求され,その材料は製造面での重要なノウハウになっています。棹の復元は光学的に検出され,サーボ増幅器で復元に要した電流量が表示されます.電流量の計測は随分精密に行えますから,電子はかりがあまり力の要らない微量,超微量の測定を最初に目指したのは当然かも知れません。この頃有機元素分析でクロマト分離法が検討され始めましたが,3~5 mgの試料を燃焼させるとクロマトピークの高さと成分量が比例せず,試料量を1 mg以下に制限することが求められました。この試料量の採取には石英トーションはかりを使うことも考えられたのですが,操作が面倒で時間がかかり,たまたま出来たばかりの手軽な超微量電子はかりに期待が持たれました。アメリカのCahn Electrobalance 26がまず名乗りを上げ,以来同類の製品が次々と市販されるようになりました。今日電子はかりと言うと数百グラムを測る化学はかりのことのようになっていますが,それはパワートランジスタによる大電流の制御が出来るようになってからのことで,もともとは微小電流の測定による微量,超微量はかりの必要性がきっかけになっています。
電子はかりも初期には棹の水平の復元という伝統的なメカニズムに拠っていましたが,間もなく荷重を載せた皿を電磁的に持ち上げ,その電流を測る新しい機構の電子はかりが考案されました(図4)。
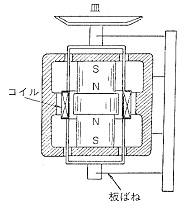
図4.電磁式上皿はかり
しかし皿の中心位置を動かさないためには,可動部を極めて柔軟な支持体で留めなければならないので,その材料に苦心が払われました。最大荷重200 g,読み取り限度0.1mgの化学はかりでは,皿の支持体の構造や材料面で実現が容易なので,まずスイスのMetler社やドイツのSatorius社から大量の電子はかりが製造販売され,このため従来の機械式はかりは化学の実験室から次第に姿を消しました。電子はかりはその後支持体の研究と高感度の電子部品の進歩で微量化が進み,やがて最大荷重2 g,読み取り限度1μgの微量はかりの領域に広がり,近年は超微量はかりと称して0.1μgを表示するものまで出来ています。棹の回転という機構を排除して,直接微小な質量変化を重力で検出するには,支持体の動きも極限の柔軟性を必要としますが,その支持法や材料については各メーカーにノウハウがあり,詳しいことは公開されていません。
超微量電子はかりの読み取り限度は0.1μgになっていますが,実際には表示値のばらつきがあり,はかりの個体差や環境条件にもよりますが,標準偏差は0.3~0.5μgあたりが多いようです。それでも0.5 mg前後の超微量試料の計量に用いて一応定量分析の要求精度を満たすことが出来ます。超微量電子はかりは手軽で便利ですが,振動や温度,空気の流れには敏感で,除振台や空調機,風防シェルターなどの配慮は微量電子はかりよりさらに厳しくしなければなりません。人体の発する熱線や呼気も影響が大きく,あまり顔を近づけて測定するのは危険です。はかりの電源部品から発する熱はゼロ点移動につながるので,最近は微量はかりでも超微量はかりでも電源部と測定系のハウジングを離して設計するメーカーが増えています(図5)。

図5.微量(超微量)電子はかり
はかりのルーツから石英トーションの超微量はかりと超微量電子はかりの二種に別れますが,分析の実務では超微量電子はかりに軍配が上がります。読み取りがパネルの表示で出来,プリンタに印字したり,コンピュータにデータ入力できる現代性があります。ただし電気量を質量に変換しているので,時々標準分銅を用いて表示を校正する必要があります。一方石英トーションは電子はかりより遥かに精密な測定を行いますが,測定に時間がかかり,表示が今のところ電気的ではないのでテータ処理が出来ず,当面は超微量分析法の究極の可能性を求める研究用の測定装置と言えましょう。
電子はかりの校正については,最大荷重の2 g校正用分銅を皿に載せて,分銅質量とはかりの表示を一致させればよいのですが,これだけではスパン全体での質量対表示値の比例性が保たれているかどうか分かりません。途中2~3箇所でも同様の検定をして比例性の確認をしますが,一般の分析室には校正用分銅セットが備えてないと思われますので,メーカーの定期検査のときに校正を依頼しなければなりません。石英トーションはかりでは表示値がトーション糸の回転目盛りになりますので,標準分銅で1目盛り当たりの質量,すなわち感度係数を検定することになりますが,質量対目盛りの直線性は極めてよいので,あとは未知物体の測定で得た目盛り数に感度係数を掛ければ真の質量を知ることができます。
3.サンプリング
超微量分析の試料は極めて僅かですが,試料ボートなどに入れて量るので,案外重い物体がはかりの皿に載ります。微量分析には規格にある1gの白金ボートがよく使われますが,荷重が大きいと測定値のばらつきも増え,またボートの表面積に応じて吸着水分の量なども増大するので,超微量分析にはもっと小型,軽量のものが必要です。規格品はありませんので,白金板を切って自作しなければなりません.横型の燃焼管に向いたものの他,目的によっていろいろな試料採取容器が考えられています6)。
図6-aでは厚さ0.02 mmの白金箔を10×4 mmの大きさに切り,型で押して25 mgのボートを作っています。6-bでは耳のある円板を切り,耳の方向に折り目を付けて半円を上に曲げ,もっと軽いボートにしていますが,こぼれ易いので微粉末以外は不向きでしょう。8×2 mmの石英毛管に細い足を付けた6-cの試料採取管は40 mgの質量になりますが,燃焼管や分解液に入れて処理することが出来ます。試料採取後は立てて置くのがよいので,スパイラルのホルダーに入れるか,穴のある金属台に立てます。試料採取管を使って別の容器に試料を移すときは,融点測定用の毛管6×1.8 mmを切り,一端を閉じて30×0.5 mmの足を取り付けると25 mgの試料はかり管が作れます。試料を入れて計量し,ピンセットでつまみ,容器に軽くタップして落とし,もう一度計量してその差を試料量とします。白金や石英製の試料採取器具は濃硝酸で洗い,高温で焼いて保存します。
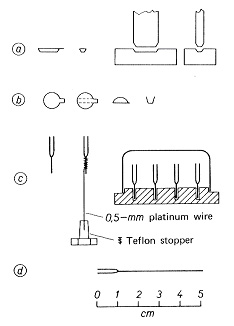
図6.サンプリング容器
アルミニウム製の場合はウレタンフォームの板に金型を押し付けて簡単に作れます。熱には弱いので加熱できませんが,水→アセトンで洗い,150℃で乾燥します。白金や石英は繰り返し使えますが,実試料では汚れも蓄積しますので,様子を見て洗浄を行います。
超微量分析のサンプリングやその他の操作ではデリケートな動きが必要なので,ピンセットなどでは手の振動が伝わって失敗することがあります。外科手術用の切除かんしを用いると手の動きが減少して確実になります(図7) 7)。
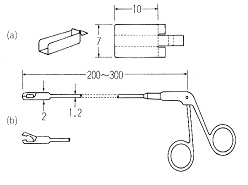
図7.切除かんし
一方の手ではさみのように動かして物を掴みますが,先のほうをもう一本の手の人差し指に載せて操作します。色々な寸法のものがありますので,目的によって選びます。細い管の奥まで物を入れたり出したりする時には重宝な道具です。
はかり取った試料が吸湿したり,揮発したりすると試料量自体が変化しますので,対策が必要です。試料量が微量になるほど単位量に対する表面積が増えますので,この影響は大きくなります。特に近年オートサンプラーに多数の試料を格納して,自動的に順次分析処理する方式が増えていますが,順番が回って来るまで長い時間がかかるケースがあります。アルミニウムカプセルに密封するのが有効ですが,走査熱量計に付属のプレス器を利用するか,もう少し安価で簡易なものもあります。アルミニウムの融点は660℃ですから,燃焼管などに入れると融けて破壊し,酸化アルミニウムの粉末になります。液体試料の超微量サンプリングはさらに困難になりますが,一端を閉じた外径約1 mmのガラス毛管を計量してピンセットで持ち,ガスバーナー上で温め,すぐ一滴の試料液に開口部を入れて1~2 mm試料液を引き込みます。手回し遠心器のガラス管に入れ,試料液を毛管の底に集め,開口部をガス炎で閉じて冷後計量します。分析に先立って毛管を折り,燃焼管などに挿入します。
4.超微量分析への道標
微量領域のCHN分析はすでに電子化,自動化が進んで機種も豊富に揃っていますが,これをそのまま超微量領域に使って満足な機能を発揮するかはまだ問題が残っています。その上提供できる試料も貴重で僅かなものが多いと考えられるので,失敗は許されません。検出器の感度をさらに高くし,ベース信号を極限まで小さく安定させる必要がありますが,両立は技術的に困難な要求です。それでも研究機関の要望によって,現在の機種のまま動作条件を設定して超微量分析への利用を目指す地道な検討が行われています。試料量をどこまで小さく出来るかはこれからの課題ですが,超微量電子はかりで試料を計量し,装置を注意深く安定させれば,500~200μgあたりまでは対応できそうです。
微量領域のCHN分析計は熱伝導度セルを検出器に使ったものが一般ですが,成分濃度と信号強度はよい比例性を持っています。この検出器でモル濃度を横軸に,信号強度を縦軸に取って検量線を描くと,モル分率で5%あたりまで直線ですが,それ以上になると水平化の方向に曲がってきます。微量分析では1~3%くらいで測定することが多いと思いますが,超微量分析では1%以下になり,相当な希薄成分の検出をしなければなりません。希薄であるからと言って微量分析に劣る分析結果を出しては技術者としての責任が問われますから,ここは工夫で切り抜けるしかありません。1%以下の希薄成分の検量線になると,不明の理由で少し曲がっているケースが多くなります。前回分析した試料の残像やキャリヤーガスの不純分などが考えられますが,曲がりがある程度再現する状態では検量線を直線とせず,2次または3次式の曲線であてはめると良い結果が得られるようです8)。ただしこの検量方式では与えられた装置の条件で複数の標準試料を繰り返し分析してデータを集め,どの式が統計的に最も誤差を小さくするか試行錯誤をしなければなりません。最小二乗法を取り入れたコンピュータ計算によって高次の計算式が導き出されるので,これで微量分析とあまり変らない分析結果が得られると言われます。
微量分析の目的で製造されたCHN分析装置をよく調整した上,超微量領域で使用するのは新しく装置を開発するより手近で,当面はその技術を推進することに賛成ですが,要求がさらに微量化すればいずれ限界が来ることを覚悟しなければなりません。燃焼系や測定系を機械的にスケールダウンするのも方法ですが,装置の構成を考えると製造コストは現在の微量分析用と変らないので,わざわざ限られた市場のために超微量分析専用の装置を出そうというメーカーも当分は無いでしょう。そうなると現在の装置に捉われず,目標に向かって手作りの道具を考えて可能性を探ることが先決です。過去においてもそうでしたが,少々不便でも実験室で組み立てられるものは一度試してみて,状況を確かめるとその後の知恵が湧いてきます。華やかな機能を見せる電子装備のCHN分析装置も,実際に動いているのは燃焼管や検出器の中の化学物質で,化学反応を導く方法は極めて基本的な道具建てで行われています。超微量分析を極限まで迫る方法論については,電子装備に拠らず簡単な器具で既に幾つかの先駆的な実験があります。まだ幼稚に見えるかも知れませんが,将来思わぬ発展の礎となるかも知れません。
5.密閉反応器
扱う量が少なくなると容器や試薬からの汚染,反応系での吸着,測定系でのベース信号の変動などが大きく影響します。微量分析では試料量に比べかなり大量の燃焼触媒を使ったり,クロマト法を利用するものでは基本的に吸着を経過するので,超微量の領域ではいろいろな影響が予想されます。試薬汚染については固体の触媒や液体の分解液からのものが多く,この点高純度の気体には汚染物質が殆どありません。1943年英国バーミンガム大学のベルチャー教授(R. Belcher)らはCH分析に燃焼触媒を用いず,代わりにやや大きな空間をもつ石英管で酸素のみによる燃焼を行いました(図8) 9 , 10)。
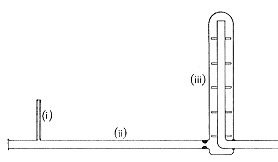
図8.Empty tube method
隔壁を設けて滞留時間を延ばし,完全酸化を達成しています。キャリヤーガスを使ってはいますが,滞留空間は一種の密閉反応器と考えられ,ここで完全燃焼を期待しています。微量分析用の装置でしたが,”Empty tube method”と言われ,吸着や空試験値のない分析法として関心が持たれました。ただ炉体の設計が複雑で実用はされませんでしたが,考え方として有効なステップでした。
酸素を充たした容器の中で試料を燃焼させるフラスコ燃焼は,ハロゲン,硫黄分析の目的で1955年シェニガー(W. Schoniger)によって考案されましたが,密閉反応器の典型です11)。試料を包む小さなろ紙以外からの不純物が無い特長がありました。またろ紙にも”Ash free”という規格品ができ,微量分析では妨害になるほどの灰分はありません。しかし超微量分析となると安心とも言えないので,ポリエチレンのシートを燃焼材に用いた小型の燃焼フラスコが推奨されています6)。これは上述のEmpty tube methodを提案したベルチャー教授が1966年発表した方法ですから,同じ人の発想は連続しています。ポリエチレンのシートは袋状のものが何処にでもありますが,不純物が含まれていているのでポリエチレンの管の一端をガスの炎で閉じ,溶けた状態で空気を吹き込み風船を作ります。0.01 mmほどの厚さのものを15 mm角に切ると2 mgほどの質量になりますので,これをろ紙の間に挟んで保存します。使用するフラスコは長さ40 mm, 径30 mmの卵形で,摺り合わせ栓の下端に取り付けた白金網に試料を包んだポリエチレンシートを挟みこみ,これに短い木綿糸を火口に取り付けます。吸収液と酸素を入れ,木綿糸を着火して蓋をはめ込み,あとはフラスコ燃焼のプロセスで処理します。
密閉容器の中で燃焼させる方法をさらに徹底させたものが封管法です。シェニガーのフラスコ燃焼法が発表されたすぐ後,1956年にはスエーデンのキルステン教授(W. Kirsten, 図9)が耐熱性のガラス管に超微量試料を採取し,銅網の短い筒とハロゲン,硫黄の除去に過マンガン酸銀を入れ,管内に酸素を充たし封管を作りました12)。数本の封管を電気炉で1時間加熱し,燃焼と同時に窒素酸化物を銅網で窒素に還元し,また管内の酸素を銅で除去します。封管の中には二酸化炭素,水,窒素が低圧で閉じ込められていますが,試薬と言えば燃焼に必要な量の酸素と銅網,少量の過マンガン酸銀だけで,しかも炉の中で長い燃焼時間がありますから,反応は定量的に進んでいます。

図9.W.Kirsten教授
冷後この封管を図10のような大きな水銀ビュレットに入れ,ポンプで真空に引いた上,封管を捻って破壊します。装置と操作法は本解説記事の「2.フラスコ燃焼法と密閉反応器」の中でやや詳しく説明していますので,参照してください。ただ水銀を大量に使うので当時はまだあまり問題にされませんでしたが,現在ならとても許されないでしょう。

図10.水銀ビュレット
たまたま1961年筆者はキルステン教授(当時助教授)の研究室に留学し,超微量分析の手ほどきを受けましたが,まずRodderUltramicrobalance Model Eによるサンプリング操作から始まり,次に封管法による燃焼と水銀ビュレットの測定を毎日練習しました。原理的には良く出来た方法で,さすが世界最先端の技術と尊敬の念を禁じられませんでした。しかし何分床から天井に達するほどの巨大なガラス器具の組み立てで,ビュレットの目盛りを読むのに梯子を二三段上がって拡大鏡を覗くという作業が必要でした.超微量の試料を扱うにしては道具立ての方がかけ離れた大きさで,その上一日中立ったままでは体も疲れるし,何とか実験台の上で作業が出来ないものかと思案しました。
「窮すれば通ず」と言いますが,無い知恵を絞っている内に思いついたのは,燃焼用の封管を作って水,二酸化炭素,窒素だけを小さな空間に閉じ込めたのですから,このまま封管をビュレット代わりに使えないかと考えました。それまでは巨大なビュレットを完全真空にするだけでも大変で,測定は午前中に一回,午後二回がせいぜいでしたから能率はすこぶる悪いと言わざるをえません。そこで取りあえずはCHN同時分析まで欲を出さずに,窒素分析だけを試みることにしました7)。概略のことは図11で分かると思いますが,封管用ガラスの一端を針状に引き,試料と銅網を入れ,他端を10 cmほどの長さに毛管状に引きます。細いステンレス製の管を通じて酸素を吹き込み,最後に毛管を閉じます.封管は700℃の炉で1時間加熱します。
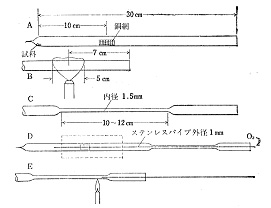
図11 封管の作り方
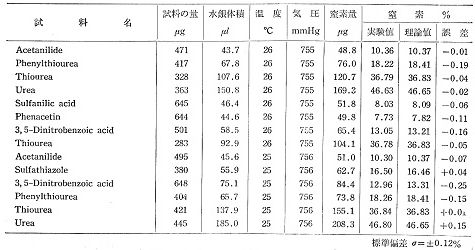
燃焼後針端を下に向けて50%水酸化カリウムの液を入れたビーカーに突き立てますと,針端は破壊されカリ液が管内を上がってきます。ハロゲン,硫黄など酸性ガスは吸収され,窒素ガスが毛管上部に集まりますから,メニスカス付近でセロテープを巻き,横に置いてメニスカスの位置をカッターの刃で記録し,毛管を切り取ります。マイクロシリンジでカリ液を洗い出し,乾燥させてから空のガラス毛管を化学はかりで計量し,次にマイクロシリンジで水銀を窒素メニスカスまで入れ,もう一度計量します.窒素ガスの体積が水銀の質量に置き換わっていますから,随分正確な測定になります。水銀のメニスカスとカリ液のメニスカスは同じ形状ですから,殆ど誤差はありません。ただ少量ですが水銀を利用するので,作業はペトリ皿の上で行うなど注意が必要です。この方法で300~600μgの試料について表1の分析値が得られましたが,標準偏差で0.12%となり,微量分析よりむしろ精確な結果となっています7)。
表1.Decimilligram 窒素分析例
この方法はその後CHN分析にも発展しましたが,操作はやや面倒で,可能性を探求したのに止まりましたが,この経験は後年になって封管燃焼の特長を生かしたハロゲン分析に活路を見出しました13)。試料の燃焼は窒素分析と同じで,一端を閉じたガラス管に試料を入れ,酸素を充たして他端を針状に引き閉じます。一時間燃焼後,吸収液を入れた小ビーカーに針端を突きたて破壊します。管内は少し減圧になっているので,吸収液は1~2 cmの高さまで吸い込まれます。約30分放置するとハロゲン種は定量的に吸収液に溶解しますから,図12のように細いテフロン管で洗浄液を噴射し,内部を洗い出します.吸収液としては塩素の場合60%イソプロパノールを用いますが,臭素の場合は亜硝酸ナトリウム水溶液,またヨウ素の分析には2%ヒドラジン水溶液を用います.最終的には0.0005 mol / Lの硝酸銀標準液で電位差滴定を行います。
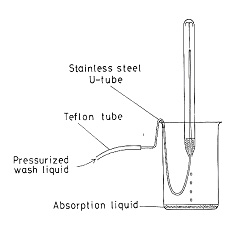
図12.封管の洗浄
面白いのはフッ素の分析で,封管のケイ素源と反応して四フッ化ケイ素を作り,フッ素のイオン化が困難となります.そこで当量より少し多い炭酸ナトリウムの粉末を封管に入れ,燃焼すると全部がフッ化ナトリウムとなり,容易にイオン化できます14)。ただ燃焼温度を高くすると炭酸ナトリウムが溶融してガラスに侵入し,一緒にフッ素も失われます。580℃が無難な温度として推奨されています。燃焼管内部を水で洗い出し,ランタン-アリザリン試薬で吸光光度法により定量できます。テフロンなど分解困難なポリマーも適用可能です。
キャリヤーガスを使う方法では流路が長くなったり,燃焼系で滞留時間が必要なことから装置規模が大きくなる傾向があります.そこで密閉容器並に小さくまとめる工夫が提案されています6)。図13では石英の管にPt―Rhのスパイラル発熱線を取り付けたキャップをはめ込み,下から試料ホルダーを入れ,酸素を通じておきます。スパイラルに規定の電流を加えると白熱し,試料は瞬間的に燃焼します。酸素はスパイラルの上下から流れて来るので成分が逃げることはありません。燃焼ガスは短い燃焼管に送られ,白金触媒で酸化を完結します。この後燃焼ガスはクロマト法によってCHN分析,吸収液に導いてハロゲン分析が可能です。またキャリヤーガスを水素に変えると,硫黄を硫化水素として吸収液に補集し,ヘリウムに変えると白金炭素を用いて酸素分析も可能となります。

図13.超微量燃焼装置
6.湿式分解と滴定
ケルダール分解法は1883年デンマークの農芸化学者ケルダール(J. Kjeldahl)によって考案されましたが15),ジュマ法より遥かに古い分析法です.濃硫酸で有機窒素をアンモニウムイオンに変換し,強アルカリを加えてアンモニアを蒸留,塩酸標準液で滴定しますが,アミノ態窒素に有効な方法ですから,農学,薬学,医学方面で古くから使われてきました。しかし微量分析から超微量分析となるとアンモニアが弱塩基であるため,当量点付近でのpH変化がますます緩やかになって終点が見分け難くなります。
アンモニアを窒素ガスに酸化する試薬としては強アルカリ性次亜臭素酸ナトリウムがあり,溶液から窒素を気泡として分離することが出来ます。希薄溶液からでも窒素気体として集めるので超微量分析に向いています。この方法は1937年金沢大学の故岩崎憲教授が若いころ医化学研究のためドイツ留学中に考案されたもので,その後アゾトメトリー(Azotometry)の名が付けられました16)。
2NH3 + 3BrO– → N2 + 3H2O + 3Br–
アゾトメータの形状は図14のようにジュマ窒素分析のものとよく似ていて,遮断球があり,下部には反応室とさらに活栓があります。始めにアゾトメータを倒立させて遮断球に食塩水を吸い込み,次に元の位置に戻して下部を試料液に挿入し,上から減圧によって反応室に吸い込みます。試料液は洗液によってもう一度吸い込まれます。この後二酸化炭素を下部から通じ,計内の空気を追い出し,さらに強アルカリ性次亜臭素酸ナトリウム液を上から吸い上げて活栓を閉じます。アゾトメータを振ると二酸化炭素は吸収され,窒素ガスが低圧で残ります。食塩水に下部を挿入し,活栓を開くと窒素ガスが目盛り管に集まり,体積が測定されます。
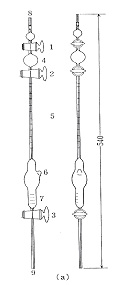
図14 アゾトメータ
原理的にはケルダール分解とジュマ窒素体積法を組み合わせたものですから,分解用触媒や体積に関係する気圧,温度,目盛り管内壁の濡れなどの補正は当然考慮しなければなりません。またアミノ態窒素以外の測定も出来るので,例えばニトロ化合物,芳香族アミン,ヒドラジンなども化学的前処理によってアゾトメトリーの対象となりますから,応用域は広範です。これらについては文献を参照してください2, 16).岩崎教授の名前が出たので書き留めておきますが,われわれが実験室でしばしばお世話になるガラスフィルターは,同教授がドイツ留学中リンの定量をするのに作ったのが最初と言われます。リンを含む試料をケルダール分解し,モリブデン酸アンモニウムを加えてリンモリブデン酸アンモニウムの沈殿として濾し分けようとしとところ,沈殿が細か過ぎてろ紙の目を通り抜けてしまい,代わりにアスベストでは目が詰まってろ液が通らないので,ガラスの粉を半融した円板をガラス漏斗に融着し,アスベストの面積を増やしてろ過したところ効率よくろ過が出来たそうです。このガラスフイルターは後にエナガラス社の製品となり,いまでは世界中の化学実験室で無数に使われていますが,誰が発明者なのか知られないままになっているのは残念です。
ハロゲンの定量では燃焼管法より酸素フラスコ法が優勢になりましたが,間もなくイオンクロマト法が普及し始めて再び燃焼管法と組み合わせた装置構成が試みられるようになりました。まだ最終的な評価はされていませんが,微量分析の領域では受け入れられる分析精度と報告されています。しかし超微量分析となるとイオン交換樹脂の脱吸着やセンサーのノイズなど未知の関門があり,素直に目的を達するかどうか検討が必要です。現状では燃焼管法でもフラスコ法でも分解ガスを吸収液に捕集し,精度の高い滴定法で成分の測定をするのが無難のように思います。ハロゲンイオンの定量には1959年ジフェニルカルバゾンを指示薬として硝酸水銀標準液で滴定する方法が提案され17),終点認定の明確さで最も精確とされました。この方法はJISや薬局方でも採用され,超微量分析にも応用されかかったのですが,その後水銀試薬の全面禁止でこの方法は使えなくなりました。JIS,日本薬局方では水銀滴定を止めたあと,銀電極と硝酸銀による電位差滴定に切り替わっています。
電位差滴定法は自動記録装置のついた微量分析向きの市販品がいろいろあって,滴定終点を数値で指示しますが,普通微分信号の極大になったところを教えます。当量点付近で電位変化が急でないと精確な判定ができませんが,ハロゲンのうち塩素イオンは当量点に達する前から塩化銀の解離があり,臭素イオン,ヨウ素イオンに比べ電位変化がなだらかになります。
AgCl ⇔ Ag+ + Cl–
微量分析では試料液にエタノールやイソプロパノールを添加して解離を抑制していますが,超微量分析では沈殿物に比べ液量が多いためイオン化が進んで測定が困難になります。このため試料液の量を少なくしてイオンの希薄化を防ぎ,指示電極に双白金電極を用いる電流滴定法が提案されています。
装置の例を図15に示しますが6),滴定槽には2mlの酢酸を入れ,図13の燃焼装置で燃焼分解した試料ガスを下から通じると塩素は吸収されて塩素イオンとなりますから,かき混ぜながらC5の2本の白金線からなる双白金電極に分極電圧を加え,電圧を記録します。図には描かれていませんが,滴定槽の向かい斜めの方向に入り口があり,0.0002 mol/Lの硝酸銀を自動ピストンビュレットで加えます。
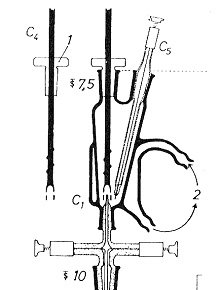
図15.超微量双金属電位差滴定
当量点付近で急な電圧のピークを生じますから,その最高点を滴定終点とします。電流量によって多少ピークの鋭さが違ってきますが,抵抗器の加減で最適の電流を探します(図16)。臭素やヨウ素の定量では燃焼によって臭素酸やヨウ素酸を生じるので,キャリヤーガスを水素にすることが提案されています。生成した臭化水素やヨウ化水素は酢酸に捕集され,塩素イオンと同じ方法で滴定が可能です。超微量滴定に必要なマイクロビュレットは一般的にマイクロメータをガラスシリンジに取り付けたものが多いのですが,精密ではあるもののまだ手動が多く,モーター駆動の自動ピストンビュレットは多くありません。ビュレットを取り替えることで500~50μL容量のものがありますが,滴定精度は標準偏差で0.1~0.05μLと言われます。
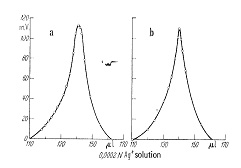
図16.分極滴定曲線
a:R=1011ohm B:R=109ohm
双白金電極の動作原理は少し込み入っていますが,簡単には両極を+と―に分極させ,当量点までは塩素イオン,それ以後は銀イオンが存在してそれぞれの電位を打ち消し,僅かな電流が流れますが,当量点では何れのイオン濃度も極小となりますから,電流も極小値を示すことになります。電極間を電位差計で測っていると,図16のように当量点で極大値になります。双白金電極は細いガラス管の先に装着でき,また参照電極を使いませんので,微量の試料液の測定に向いています。電極表面は滴定を繰り返すとハロゲン銀の膜で覆われますが,厚くなるとそれ自身で銀イオン応答をするようになり,滴定曲線の障害となります。チオ硫酸ナトリウムの水溶液に漬けてハロゲン銀を溶解除去するか,もし電極が白金線の場合は細かいサンドペーパーで擦ればすぐ快復します。
フッ素はハロゲンの一種ですが,塩素や臭素とはかなり化学的性質が違っていて,分析方法も全く異なってきます.フッ化ランタンの単結晶を円板状に切り出したフッ素イオン選択性電極がありますが,微量分析では燃焼分解したあと,吸収液にフッ素イオンとして捕集し,メスフラスコで定容した試験液のフッ素イオン濃度を電位差計で直接測り出すことができます。しかし濃度の対数が電位差に比例するので,僅かな電位差の測定誤差がイオン濃度の認定に大きく影響します。高感度の電位差計を用い,周到な静電遮蔽をして測定を行います。しかし超微量領域になるとこの方法はもう利用困難でしょう。フッ素イオン選択性電極をセンサーに用いて,硝酸ランタンによる電位差滴定を試みた人もありますが,ランタンが3価の希土類で,LaF3を形成する反応ですから滴定値が上がらず,精密な定量はできません。
微量から超微量にかけ有効に利用できる方法は,アリザリン錯体による吸光光度法です。ここではフラスコ燃焼法で数mgの試料を分解し,吸収液を100mLのメスフラスコに定容し,そのうち1~30μgのフッ素に相当する量をピペットで取り,50mLのメスフラスコ中でアリザリンコンプレキソン-ランタン試薬(同仁化薬:ドータイト-アルフッソン)と発色させ,620 nmの波長で吸光度を測定します2, 18)。微量分析でも試料中の一部のフッ素を取り分けて利用するので,超微量分析では全部を使って丁度よいことになります。予めフッ化ナトリウム標準液で作成した検量線を使って試料中のフッ素量を知ることが出来ます。ただ発色試薬自身がすでに赤ぶどう酒色をしているので,フッ素と結合して僅かに青みを帯びますが,感度のよい分光光度計を用いなければなりません。
硫黄化合物は燃焼によって三酸化硫黄と二酸化硫黄を生じますが,スルファニルアミドやスルフォン酸など分子内で酸素と結合しているものは前者を多く,スルフィドやチアゾールなど酸素と結合していないものは後者を多く生産するようです。三酸化硫黄は水に溶けて硫酸イオンとなりますが,フラスコ燃焼法では吸収液に過酸化水素を少量加えてあり得る二酸化硫黄を硫酸イオンに酸化してから定量しています。エチルアルコールやイソプロピルアルコールなど有機溶媒を加え非水溶液とし,指示薬にトリンを添加して過塩素酸バリウムで滴定というのが微量分析では早くから行われましたが19),当量点で薄い黄色からピンク色に変化するのが微妙なので,最近はカルボキシアルセナゾを指示薬に使い,赤→青の変化点を終点とするのが推奨されています 2, 20)。
微量分析で使う指示薬滴定法は,原理的には超微量分析にも応用できると思いますが,液量が少ないと変色も見分け難くなります。そこで発想を変え,ハロゲンの定量に用いた図15の双白金電極で終点の検出が出来ないか検討が行われました。このため試料の熱分解に水素を用い,硫黄を硫化水素に変換してアルカリ液に吸収させ,硝酸銀で滴定する試みが提案されています6)。
S2- + 2Ag+ → Ag2S
前出の図13の分解装置でキャリヤーガスに水素を用い,100μg以下の試料を分解し,滴定槽に入れた500μLのアルカリ液に硫化水素を吸収させます。双白金電極を入れ分極電流を流し,両極の電位差を記録します。0.005 mol/Lの硝酸銀標準液をピストンビュレットで加えると,ハロゲンの場合と同じように当量点で電位差極大を示しますから標準液の注加量を記録します。
7.おわりに
科学技術は常に過去を学びながら次の発展を目指します.分析化学も錬金術から道具や薬品の知識を深め,グラム単位の試料を用いる技術の体系を作りあげました。沈殿による分離法,試薬による呈色反応,電位差測定によるイオンの計測などは戦前まで分析法の中心でした。自然界にある多彩な物質の究明に分析化学は大きな貢献をしましたが,究明の対象が普遍的なものから特殊なものに広がると,微量な物質の扱いや複雑な構成の中から目的物を選別して定性,定量を行う技術が必要となりました。有機物の微量分析法は20世紀の歴史的な遺産ですが,内容的には前半の手分析から後半の電子機構を援用した自動分析に成長しました。時期を同じくして吸光分析法,クロマト分離法,原子吸光法,イオン電極法,質量分析法など高度の機器分析法が発展し,これらの一部が微量分析法に取り入れられています。分析実務に関しては昔考えられなかったほど便利に,迅速に,精確にデータが得られるようになりました。
さて21世紀が始まった現在,微量分析は次にどう進むべきか議論がされています。手近な作業として現在の微量分析法をもっと高精度のものに仕上げることがあります。データのばらつきは標準偏差で評価されますが,過去の±0.3%というのは少し甘すぎるので,状況にもよりますがもう少し厳しい方向に行ってもよいでしょう。現在は限られた数の元素しか測定対象になっていませんが,有機合成の研究ではどんどん新規の元素を構造に取り入れて機能性を拡大しています。今まで対象としていなかった元素の定量が今後要求されるのではないかと思っています。分析法が電示的になりすぎて,データ処理が便利になった反面,データから直接成分の含量を求めることが出来なくなっています。標準試料で感度係数を求めるしかありませんが,標準試料の信頼性にいささか疑問が持たれるケースもあり,今後製造,保全,検定,供給態勢などを整備する必要があります。
分析試料の規模についてはマクロ化とマイクロ化の両方に残された課題があります。微量分析法が便利で普及しすぎて,皮肉にも昔あったマクロ分析に戻るのが困難になっています。しかし食品,飼料,工業原料,燃料など品質検査には欠かせない分析項目で,今後も分析法の改良と機器化は進めなければなりません。最後に辿りつくのは分析法の超微量化です。すでに20世紀の後半この領域を目指して散発的な研究があちこちで行われましたが,まだ定着した分析法に育ってはいません。マニュアル分析のものが殆どですから,電子化された微量分析用の装置に馴れた方はすぐには食指が動かないでしょう。出来上がるまで待つというのも賢明な行き方ですが,作り上げるのに参加しようと言う方もあって欲しいと思っています。小さな発案でも記録に残してもらえば,また別の人が取り上げて更に改良してくれるはずで,多くの方々の協力が欲しいところです。フラスコ燃焼のシェニガー博士の研究室を訪ねたとき,どこにもフラスコが見当たらないので聞いたところ,世界中で私の方法の改良をしてくれるので,自分は他のことをやりますとのことでした。21世紀をどのように導くか現役の方々に期待を寄せていますが,得意の領域で何か足跡をのこして下さるよう願っています。
8.参考文献
1) F. Pregl: “Die quantitative organische Mikroanalyse”, Springer, Wien (1916).
2) 日本分析化学会有機微量分析研究懇談会編:”有機微量定量分析”,南江堂(1969).
3) P. L. Kirk, R. Craig, J. E. Gullberg, R. Q. Boyer: Anal. Chem., 19, 427 (1947).
4) 穂積啓一郎: 分析化学,19, 163 (1970).
5) 山本健太郎編: “質量の精密測定マニュアル” ,日本規格協会(1981).
6) G. Tolg: “Ultramicro Elemental Analysis”, Wiley-Interscience, New York (1970).
7) 穂積啓一郎: 化学の領域,17, 873 (1963).
8) 佐藤綾子: 私信(静岡県立大学薬学部).
9) R. Belcher, C. E. Spooner: J. Chem. Soc., 1943, 313.
10) G. Ingram: “Methods of Organic Elemental Microanalysis”, p20, Chapman & Hall, London, (1962).
11) W. Schoniger: Mikrochim. Acta, 1955, 123.
12) W. Kirsten: Mikrochim. Acta, 1956, 836.
13) Y. Tanaka, A. Okazaki, K. Hozumi: Mikrochim. Acta, 1991, III, 169.
14) Y. Tanaka, A. Okazaki, K. Hozumi: Microchem. J., 43, 62 (1991).
15) J. Kjeldahl: Z. anal. Chem., 32, 236 (1883).
16) 岩崎 憲,大月 理,中島誠一: 十全会雑誌,42, 2125 (1930).
17) F. W. Cheng: Microchem. J., 3, 537, (1959).
18) M. E. Fernandopulle, A. M. G. Macdonald: Microchem. J., 11, 41,(1966).
19) J. S. Fritz, S. S. Yamamura: Anal. Chem., 27, 1461 (1955).
20) K. F. Novikova, N. W. Basargin, M. E. Jsyganova; J. Anal. Chem. USSR, 16, 395 (1961).