



TOPICS
CHNフォーラム
第五部 分析値の信頼性
- 1-1.はじめに
- 2-1.測定値のばらつき
- 3-1.サンプリング誤差
- 3-2.CHN分析計の誤差
- 4-1.精度と真度
- 4-2.分析誤差と不確さと
- 5-1.検量線と回帰分析
- 6-1.おわりに
- 7-1.参考文献
- 関連装置
1-1.はじめに
定量分析は目的成分の含量率を正しく言い当てるための化学技術ですが,ただ一つしかない真実の値に迫ろうとしても分析過程でいろいろな不安定要素を取り込み,得た結果に100%の自信を持つことができません。有機元素分析では化学構造の予想された試料の場合,あらかじめ含有率の理論値まで出しますが,分析結果がそれに一致しないと測定上の間違いなのか,それとも試料が予想のものと違うのかと迷います。測定上の通常の間違い範囲が分かっていると,分析結果と理論値の差がその範囲内にあれば試料の化学構造はそのまま支持されますが,この範囲を超える間違いがあると試料の方に原因があるとされます。このときは試料が不純物を含んでいるか,または化学構造が予想と違っているのではないかなどの検討課題に移ります。
許容誤差という言葉が古くからあって,今でも非公式に使われていますが,分析者の立場からすると都合のよい表現です。許容誤差が広いほど理論値と分析値に差があっても苦情を言われないので助かりますが,そのぶん化学構造が少々違っていてもそのまま認めてしまう危険が増えてきます。誠実に努力しても避けられない分析誤差と,化学構造の確定に必要な分析値の精度との接点を,無数の分析例を通じて自然発生的に作り上げたものが,±0.3%の許容誤差で,プレーグル時代から現在もまだ続いています。
分析値は分析化学操作の結果得られるものですから,分析手順に沿ってランダムな間違いが次々と入ってきて結果に影響を与えます。はかりによる試料量のばらつきや,反応条件の不適正,充填試薬の消耗,反応生成物の損失,外部からの汚染,生成物の不完全回収,回収成分の測定値のばらつきなどよく知られた誤差源がいろいろあります。CHN分析計のような複雑なブラックボックスの中では燃焼系,吸収系,分離系,電気系,機械系が一体となっていて,その中で何が起こっているのか覗いて見ることもできません。誤差源はどこにでもあって実態は掴み難いのですが,誤差源どうしは決まった統計ルールによって相互に組み合わされ,結果としてばらつきを持った分析値がわれわれに与えられます1)。統計ルールそのものの基礎理論は相当古くからありましたが,最近少し見方を変えて解釈するようになり,新しい用語なども登場してきました。
許容誤差の考え方も一世紀を過ぎた現在,昔のままの±0.3%でよいのかという反省もでています。測定器や装置の進歩でもう少し狭い範囲でよいという方々がある一方,試料量を1 mgまたはそれ以下に引き下げて,なお±0.3%を維持しようと努力しているグループもあり,簡単には割り切れない面があります。また含有率の高い元素と低い元素に同じ許容誤差を与えるのはおかしいとの説もあります2)。同じ成分の分析でも測定法が違えば結果のばらつきも異なると思われますが,いくつか気になる点について考えて見たいと思います。
2-1.測定値のばらつき
的を矢で射るというのは実戦にも役立つし,スポーツとしても面白いので,昔から盛んに行われました。那須の与一が屋島の浦で平家の扇のかなめを打ち抜いた話は有名ですが,与一がもう一度やって同じように当てられたかどうかは分かりません。目標が決っていて精一杯それに近づけようとしても,複雑な要素がランダムに作用して目標を外すことは普通に見られる現象です。しかし同じことを繰り返してそれらのデータを集めると,データの散らばりには限度があり,同時に目標付近に集まるデータが多いことに気づきます。(図1)
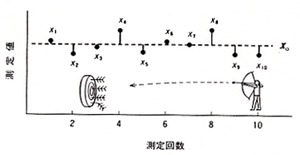
図1.測定のばらつき
標準偏差という統計用語はポピュラーに使われていますが,標準とは自然に起こる現象のことで,データの数値に人間の作為が入っていません。目標に向かって努力した結果ですから,目標付近で一番確率が高く,これから離れると確率が下がって最後はゼロになります。自然現象であれば中心から両側に向かって左右対称の減衰形確率分布すなわち正規分布曲線を描くはずで,この曲線の広がりがデータのばらつきを表します。(図2)
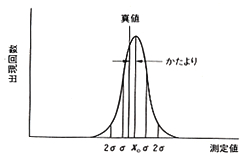
図2.正規分布曲線
分析化学のデータは割合数が少ないのが特徴です。精密な結果を出そうとするとそれだけ念入りに操作を行うので,時間もかかり,繰り返し分析をいつまでも続けられません。微量分析では提供される試料の量も限度があって2~3回のデータを判断資料としますが,実はその背後にある見えない無数のデータ群の仮の代表を取り出したに過ぎません。もう一度分析を繰り返すと違ったデータが得られますが,これも背後にある無数のデータ群の違う場所を取り出しただけで,背後のデータ群は与えられた分析条件では動かない性質のものです。
背後にある動かないデータ群は母集団といわれますが,数の少ない分析データで母集団の散らばり状況を推定するのはかなり難しいことです。微量分析では多くても10回くらいのデータで母集団を推定するのが通例ですが,それ以上になると実験中に分析条件が変わってきて,却ってデータのばらつきが悪くなることがあります。図1には測定回数とそれぞれの測定値xiを表していますが,平均値xoを中心に上下に偏差を生じています。
偏差は平均値に対して+または-の符号で与えられますが,ばらつきを評価するのに全部を合計しても+と-が打ち消しあってゼロになってしまいます。そこで偏差を二乗して正の値にしてから合計します。偏差平方和がこれですが,測定回数とともに大きくなりますので,測定回数にかかわらない値として測定回数nで割ることになっています。この値は偏差が自然に出て平均値から偏りの無い分散をしているという意味で不偏分散variance, Vと言っていますが,次の式で求められます。
V = Σ(xi -x0)2 / (n – 1)
ここでデータ数nの代わりにn – 1で割っているのは奇妙に見えますが,それは統計学でいう自由度がデータ数より1個少ないことによります1-p18)。今データがA, B, Cとあって,(A + B + C )/3 =x0と平均値が与えられると,自由に値が選べるのはA, B, Cの内二個しかありません.第三のデータは平均値x0を出したことによって決まってしまい,自由なデータが1減っているからです。ばらつきは自由に選べるデータで決りますから,この場合データ数から1を引いた値を自由度として計算します。
n – 1の自由度で割る別の利点は,得られた少数データで背後にある母集団のばらつきを推定するのに役立つことです。少数データには異常な大外れのデータが入りこみ難いので,どうしても本来のばらつきより小さいように評価してしまいます。例えばある人が車の運転で1年間無事故無違反であったとしても,一生を通じて事故率はゼロで済まないでしょう。n – 1の自由度で割るのは,分母を小さくしてばらつきの値を大きくしていますが,少数データで母集団の推定をするとき,真のばらつきにより近づける効果があります。逆にデータ数が多ければ1を引いても引かなくてもばらつきの値は殆ど変わりません。
不偏分散は偏差の二乗を集計したものから来ていますので,その平方根を求めて元に戻すと標準偏差standard deviation,σとなり,ポピュラーに使われています。
σ=V 1/2
ただしこれはデータが正規分布していることを前提にしています。このとき得られた全データの内,標準偏差σの枠内にあるものは68%です。つまり10回の繰り返し分析の結果7回近くがこの枠に入りますが,あと3回あまりはこの枠から外れることを意味します。もし標準偏差の2倍すなわち2σまで枠を広げるとデータの含まれる確率は95%ほどとなって,事実上ほとんど全部が含まれてしまいます。それでも100%ではないので,ときどきは思いがけない異常値が現れることがあります。
ここで許容誤差±0.3%というのは標準偏差σのことか,それとも2σのことか考えておかなければなりません。確率68%で±0.3%に入れるのは初期の元素分析でも何とか可能であったと思われます。しかし確率95%で±0.3%に入れることは当時の技術を考えると無理なような気がします。憶測ですが10回の繰り返し分析のうち7回ほどが±0.3%の枠に入れば良好という時代ではなかったかと思われます。
さて化学構造の決定手段がほとんど元素分析に頼られていたころ,±0.3%の分析誤差の重みを考えてみます。ステアリン酸とオレイン酸はそれぞれ代表的な脂肪酸で化学構造もよく似ていますが,後者は二重結合が一つあります。
ステアリン酸: CH3(CH2)7CH2CH2(CH2)7COOH = C18H36O2 = 284.48
オレイン酸: CH3(CH2)7CH=CH(CH2)7COOH = C18H34O2 = 282.47
ステアリン酸の元素含有率はC=75.99%, H=12.75%, 一方オレイン酸は水素が2個少なくてC=76.54%,H=12,13%です。両者は炭素値で0.55%,水素値で0.62%の違いがあります.元素分析の誤差が10回のうち7回ほどの確率で±0.3%であれば,得られた分析値から試料がどちらの化学構造か大体判別できますが,これより大きいとどちらの構造なのか次第に自信がなくなります。
もちろん当時でも元素分析と併行して,分子量の大きさを融点降下法や沸点上昇法で,また二重結合の有無と数を臭素添加法などで測定してある程度の情報を集めていましたが,最後の決め手は正確な元素分析値でした。水素1個のあるなしまでは当時の技術では無理であったかも知れませんが,技術水準と要求水準がどうやらこの時代に±0.3%あたりで妥協し定着したような気がします。
3-1.サンプリング誤差
許容誤差という言葉が長く使われてきたにもかかわらず,それが1σでの話か2σでの話か,上の議論ではまだ決着しませんが,分析実務にとってはやはりこの言葉がなくては結果報告に踏みきれません。もともと分析誤差は複数の要因が組み合わさったものですから,要因の種類とそれぞれの強度によって分析誤差のばらつきも違ってきます。目的元素や分析方法によって許容誤差は違うのが本来ですが,それでも何となく±0.3%で納得しているのは,やはり一応のけじめが必要だからでしょう。
±0.3%を誰が最初に言い出したのか調べていますが,まだよく分かりません。欧米のテキストブックや元素分析の文献には,古くから“allowable error”として±0.3%の記述がありますから世界的にも通用する数値です3)。 数値自身が少々カリスマ性を持ってしまって,これを否定するには勇気が要りますが,確率論で解釈すると多少柔軟に意味付けすることができます。
元素分析値の誤差にはかりのばらつきが関係していることはプレーグル時代から分かっていたことで,1900年代初頭プレーグルから依頼されたクールマンは微量はかりの製作に当時の精密技術の極限に迫りました。このはかりはしかし使う側にも名人芸を要求し,それぞれのはかりの固有の動きをうまく使ってよい繰り返し精度を出す人と,これが出来ないで測定値にひどくばらつきの多い人があったようです。当然分析結果にもこの個人差が現れますので,はかり操作の習熟は分析技術者の最初の厳しい関門でした。
1940年米国化学会の分析,微量化学部門で微量はかり委員会が発足し,当時多く使われていたクールマンをはじめ,ベッカー,アインスウオース,サルトリウスなどのはかりの性能を統計的に調べました4)。表1は10回のデータを集計した結果で,1 g荷重のときと10 g荷重のときの標準偏差を比較していますが,荷重の増加はそれほど影響ありませんでした。まだダンパーや投影目盛りのない時代ですから,はかりの管理の良し悪しや使用者の技術が丸出しで,1 g荷重のとき1μgなどとても信じられない良いデータもあれば,反対に20μgあたりの気の毒なものが混在しています。1940年というと第二次世界戦争が始まろうという騒然とした頃ですから,よくこの時代にこういった地味な研究会が企画されたものかと驚きますが,さすが超大国アメリカのゆとりというべきでしょう。
1945年第二次世界戦争はようやく終わりましたが,平和がもどるとドイツのブンゲ社,ザルトリウス社などから空気ダンパーや投影目盛りなどの新技術が微量はかりに取り込まれるようになり,国産品もこれに追随して,測定に個人差があまり無くなってきました。ばらつきの責任が個人の操作技術より機械そのもののほうに移ってきて,ようやく分析結果とはかりのばらつきの関係が客観的に議論されるようになりました5),6)。
当時の微量はかりのばらつきは標準偏差で2~3μgかと思われますが,分析値の許容誤差0.3%のうち,サンプリングの段階でこの分が先に削られてしまうので,複雑な分析過程からくるその後の誤差の割り当てが少なくなります。仮に中間の値として標準偏差2.5μgの微量はかりでサンプリングを行ったとして,以後の分析操作に全く誤差原因がなければ,サンプリングエラーに起因する分析誤差はいくらになるかを見積もります。
われわれがサンプリングをするときは必ずボートやカプセルの質量を量り,このあと試料を入れてもう一度量りますから,一回のサンプリングにはかりの計量誤差が二度入り込むことになります。ランダムな誤差が重なるとばらつきは広がりますが,広がりの大きさは不偏分散の和(V1+V2)となり,結果として標準偏差σ0は次のように計算されます。
σ0=((V1+V2)1/2
空のボートと試料を入れたボートは同じはかりで計量するので,
σ0= (2σ2 ) 1/2 = 21/2 σ
はかりの標準偏差が2.5μgのとき,量り取った試料の標準偏差は,
σ0= 1.41 × 2.5 = 3.53μg
試料採取量が3 mgのとき,相対標準偏差relative standard deviation (RSD)は3.53/3000=0.001177ですから,含量70%の元素定量のときには,分析誤差として70×0.001177 = 0.082%になります。
サンプリングをしただけではまだ分析操作に入ったと言えませんが,すでに0.082%ほどの分析誤差があるわけで,しかも10回のうち7回がこのこの範囲にあるという話ですから,もし9回まで入る範囲を考えるとその2倍の0.164%になってしまいます。これで分析値の許容誤差0.3%を確保するにはあと0.136%しか残っていません。これは厳しすぎるので,多分当時の許容誤差0.3%とは10回のうち7回ほどが収まる範囲であったとするのが妥当のような気がします。
しかし機械式はかりも年々精度が向上し,1950年台には標準偏差1.5~2μgあたりが一般に供給されるようになりました6)。同時にこれ以上ばらつきの大きなものは清掃,修理を必要とする一般的な認識が出来てきました。このくらいになると上の許容誤差の計算は試料量の標準偏差として,
σ0= 1.41×(1.5~2) = 2.11~2.82μg
試料量が3 mgのとき,相対標準偏差は(2.11~2.82)/3000=0.00070~0.00094ですから,含量80%の分析のとき,80×(0.00070~0.00094) = 0.056~0.075%になります。10回のうち9回まで入る範囲となると,(0.056~0.075)×2 = 0.112~0.150%ですから,はかりに起因する誤差は許容誤差0.3%の半分以下ですみます。
最近は電子式の微量はかりが普及して,標準偏差1.5μg以下のものが普通になりかけていますので,サンプリング誤差はもっと小さくなるわけですが,試料量の方も昔の3 mg以上から2 mg以下が多くなって来て,折角はかりの精度が向上したのに試料量の相対標準偏差に関しては結局あまり楽になっていません。
あまり筋の通った話ではありませんが,私流の解釈としては1900年代前半の許容誤差0.3%は10回のうち6~7回が入る範囲であって,1950年を過ぎてから10回のうち9回まで入る範囲に技術が向上したと思っています。そう考えれば許容誤差0.3%も生かせておいて,現在では9割まで信用できる範囲になったと解釈するのが妥当ではないでしょうか。
3-2.CHN分析計の誤差
いろいろな目的の分析計がありますが,原理も測定法も違うので,分析誤差の原因を全部調べその影響を知ることは困難です。しかしCHN分析計はその中でも特に利用頻度が高く,精度向上を兼ねて分析結果に響く要素をある程度調べた例があります7)。1970年の報告ですから,電子部品も今ほど高度のものでなく,データは現在の分析計に当てはまらないと思いますが,解析方法を簡単に述べます。
まず誤差の構成因子のうち,固定的なものを集計します。A)試料の計量誤差,B)検出器の出力誤差,C)気圧の変化,D)恒温槽の温度変化,E)ベースシグナルの変化をそれぞれ相対標準偏差として見積もり,それぞれを二乗して分散の形にします。炭素値についてA)~E)を合計し,分析値の分散から差し引くと残りの分散がでてきます。この残りの分散には反応の不完全,成分の洩れ,外気の侵入,流路のつまりなど雑多な原因が含まれますが,大体A)~E)の合計と残りの分散とは似たような値になりました。分析系全体としてバランスが取れていると考えられますが,分析結果はアセトアニリドやアンチピリンを標準としたとき炭素値でσ=0.1~0.2%を得ています。2σではこの倍で許容誤差を少し超えるのもありますが,かなり昔の機械ですから仕方がありません。現在の装置で同じような解析実験をするともう少しよい結果になると思いますが,相当な時間と手間がかかりますので,実行にはかなりの決心が必要です。
4-1.精度と真度
標準試料を何回か分析するとばらつきを持ったデータ群が得られますが,その平均からのばらつきを標準偏差で表し,それが小さいのを精度がよい,また平均値が理論値に近いのを正確さが高いと長年言ってきましたが,1994年国際標準化機構の品質規格改訂(ISO 5725-1)により用語や定義に少し変化がありました8)。
まず精度についてはばらつきを標準偏差で表す点で同じですが,分析条件が固定されているときは(分析者,装置,環境,日時が同じ),繰り返し精度repeatabilityと言い,普通われわれの元素分析データはこの状況のものです.しかし分析者が複数で交代に出したデータの集計とか一ヶ月の変動を含めたデータの統計などは再現精度reproducibilityとなります.繰り返し精度のことを再現性と言う方がありますが,正式には間違っています。
一方正確さに関してはばらつきのあるものの平均をとってそのデータ群の代表とさせるには少し問題があります。ばらつきが小さいときはそれでも納得できるかも知れませんが,ばらつきが大きいとたとえ平均値が理論値に一致しても正確な分析ができているとは言えないでしょう。ISOでは正確さの代わりに真度truenessの用語を設け,データからどのくらい離れた所に真の値があるかを表すことになりました。
真の値とは厳密には知り難いのですが,よく校正された分銅の質量値や標準試料の化学構造から計算される理論含有率などを参照値として代用することができます。得られた個々のデータやデータ群の平均値が真の値にどのくらい近いかが真度ですが,要するに従来のかたよりが真値を先に仮定してそれからのデータのずれを考えたのを,逆にデータがあってそこから真の値までの距離を見る積極的な姿に変えたと言えるでしょう。
さて精度と真度が決りましたが,データの質としてはどちらも必要で,これらを兼ね備えたデータは精確さaccuracyが良いと言う事になりました。精確さは自由に変化する二つの要素からできていますので,総合評価に数値的な基準を設けることは困難ですが,要求される精度と真度がどちらも高ければ精確さも良いということになります。結婚式では花嫁が才色兼備であることになっていますが,正直言って二要素の程度と組み合わせがいろいろあって,仲人はどの程度に誉めるか言葉に苦労するようです。精確さと正確さは発音が同じで暫く混乱があるかも知れませんが,正確さが使われなくなり,真度に置き換わる時がくればこの問題も解決するでしょう。取りあえず以上をまとめると,次のようになります。
精確さ:精度+真度
精度:繰り返し精度(同一条件)と再現精度(異なる条件)
真度:真値との隔たり(旧かたより)
得られたデータは母集団の中のある場所から取り出したもので,それが偶然目標の値に近くてもそのまま信用できるとは限りません。依頼分析などで報告されたデータが期待の分析値に最終桁まで一致することがありますが,本来ばらつきのあるデータ群の一番外れたところを貰った可能性もあります。場合によると分析全体が間違った標準で流れていて,その間違いが原因で実際とは違う化学構造の理論値に一致してしまうなどということもあり得ます。
標準が狂っていてはすべてが信用を失いますが,この信用を支えるのはトレーサビリティ traceability の確保です。この用語は最近よく使われるようになりましたが,由来が辿れるという意味で,平たくいえば家の系図があって先祖から自分の代までのつながりが明らかであるというのと同じです。最近の狂牛病さわぎで牛肉のトレーサビリティを表示することになったようですが,ここでは牛の種類,原産地から処理場所,流通経路まで明記させ,消費者が何を食べているのか分かるように計画しています。
はかりの分銅はキログラム原器が出発点で,100 g,10 g, 1 gと厳密な比較測定を繰り返しながら質量を下げ,最後はミリグラム分銅に到りますが,そこではキログラム原器の質量を出来る限り精密にトレースしています。この分銅を使って校正されたはかりは目盛りにしろデジタル表示にしろすべてキログラム原器の質量から来ていますから,このはかりの測定値には質量に関して高度のトレーサビリティがあるといえます。ただし分銅の汚れやはかりの感度に変化があるとそのトレーサビリティは崩れるので,ときどき分銅の比較検査やはかりの感度の校正をしてトレーサビリティを取り戻さなければなりません。
定量分析ではサンプリングから始まって,燃焼分解,成分の分離や発色,滴定などいろいろな過程が組み合わされて結果が得られますが,いずれの過程も量の基準が同じ標準に統一されなければなりません。具体的にはサンプリングに使うはかりがキログラム原器の質量をトレースしているので,他の過程も同じ流れに乗っている必要があります。
試料の燃焼分解によって得られる成分の量は反応が100%の効率で進んだときに,サンプリングと同じ質量のトレーサビリティに乗ることができます。しかし効率が100%でなかったり,成分の損失や汚染があるとトレーサビリティは崩れます。成分の分離や発色も,標準液の一定量で同じことを行って効率や発色率を知っておけば成分量が標準化されます。滴定などに用いる量器は目盛りがJIS規格で管理されていますが,元をただせば目盛りを充たす水の質量で体積が計算されているので,これはキログラム原器の質量をトレースしていることになります。熱伝導度セルや吸光度計の電気的出力もはかりで計量した標準試料で感度係数を決めますから,それで得られた成分量は間接ですがキログラム原器の質量をトレースしています。
建前としては一連の分析プロセスがすべてキログラム原器にトレーサビリティを持っていて何の問題もなさそうですが,それでも微量分析ではあまり安心できません。例えば試料の計量に分析室の微量はかりを用い,滴定標準液に試薬メーカーから買ったものを使うと,後者は試薬メーカーの持つ化学はかりで濃度や力価を決めてありますので,分析室の微量はかりと試薬メーカーの化学はかりの標準からのずれの違いが分析結果に現れます2)。
微量はかりのトレーサビリティについては過去に大掛かりな調査があり2),9),問題点の解消にむけた努力が実を結び,現在では面目を一新していますが,それでも不特定の化学はかりと微量はかりが同じトレーサビリティを持っているかと言われると多少不安があります。滴定液の濃度や力価は分析室の微量はかりで標準試薬を計量し,これを溶媒に溶かして滴定して求めたほうが標準の統一がとれて安心のように思います。
4-2.分析誤差と不確さと
誤差は分析結果と真の価との差であると概念的に言われていますが,真の価はどうして分かるのかと言うと,それは物質の融点のように大勢で決めた公定値であったり,元素分析のように分子式と原子量から求めた計算値であったりします。真の価より高い分析値には+の誤差として,低い分析値には-の誤差として表されますが,真の価がないときは勝手に+や-をつけることが出来ません。しかし真の価は無くても分析結果はばらつきを持ったデータ群から取り出した一例ですから,その価のあやふやさは範囲で表すことができます。これが与えられたデータの不確かさで,データのほうから見た真の価のありうる距離を示していて,+や-の表示はしません.ただしこれは余り厳格には守られていないようで,±の表示をするのも見られますが,丁度標準偏差の値に本来不要な±をつける人があるのと似ています。
不確かさもばらつきが原因ですから,統計原理が働きます。不確かさが単純なものであれば,旧来の標準偏差の数値で表せますが,これを新しく標準不確かさstandard uncertaintyと呼び,uで表します.もし不確かさが複数あればu1, u2, ・・・ un が累積し,それぞれの分散の合計として合成分散Zが得られますが,
Z = u12 + u22 +・・・+ un2
これから合成標準不確かさcombined standard uncertainty, ucが求められます。すなわちZの平方根がそれで,
uc = Z1/2
となります。新しい用語が出てきて戸惑うかも知れませんが,従来の標準偏差,分散の考え方と同じですから,古い用語と置き換えればさほど難しい話ではありません。標準偏差が分かっておれば,得られた測定値と真の値の差が不確かさで表現されます。
サンプリングに例をとると,空のボートを測り,試料を載せてもう一度測り,その差をとるので,2回の不確かさが累積しています。はかりの標準不確かさがubとすると試料量の合成分散はZ = 2 ub2となりますから,試料量の標準不確かさはuc = 21/2 ubです。分析結果はそれからいろいろなプロセスを通って与えられるので,実際には複雑な合成要素が絡みあっています。
ここまでの不確かさの見積もりは確率68%で行われましたが,これを確率95%に引き上げると不確かさは2倍の範囲にしなければ納まりません。これを拡張不確かさexpanded uncertaintyと称し,Uで表します.確率を広げる係数は2倍でなくてもよいので,一般的には包括係数kと呼んで,U = k ucで表していますが,普通k = 2で計算されます。これは95%確率で見積もっているので,殆ど全部のデータがこの範囲に入ると考えてよいでしょう。例えばある分析装置のデータが標準偏差0.12%で得られるとき,未知試料の分析値72.15%の不確かさ(普通拡張不確かさを取ります)は0.12×2=0.24%で,真の値は72.15±0.24%の幅の中に95%の確率で存在することを意味します。
5-1.検量線と回帰分析
成分量とそれに対応する信号量を直角座標にとり,異なるデータの数点を書き入れるとほぼ直線に並びますが,さらにデータを増やしても大体同じ直線に乗ると考えられます。多少ばらつきはあっても結局は同じ直線の上に戻ってくるのでこれを回帰直線regression line と呼んでいますが,昔はmm方眼紙に鉛筆でデータをプロットし,目分量で定規をずらせ,どのデータポイントにも最も近い直線,すなわち検量線を引きました。このやり方は余り厳しくない分析種目でいまでもよく行われています。
しかし1/1000の精度を求める有機元素分析では目分量で検量線を引くわけにはいきません。そこで最小二乗法の計算による回帰分析を行って検量線cali-bration line1-p117)を求めます。この検量線には原点を通るものと原点を通らぬものがあり,どちらかに決めなければなりませんが,元素分析では空試験値を同時に推定するため後者を求めます。(図3)
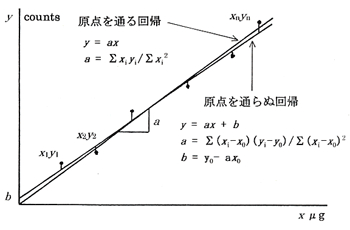
図3.検量線
成分量xを横軸にとり,シグナルの量yを縦軸にとって数点のデータを入れ,これに沿って直線を引くとこの線は勾配aと縦軸上の切片bを持った直線方程式 y = ax+b で表されます。bは正の場合と負の場合がありますが,元素分析では正の値が普通で,これを空試験値としています。真の空試験値は試料を燃焼させずに得られるシグナルですが,これと回帰分析から計算で得たものと微妙な違いがあり,実用的には後者の値を利用しています。
Y = ax+bを構成する成分量xもシグナルyもばらつきを持っていますが,どちらかが固定されないと検量線が引けませんので,この際は成分量xにはばらつきが無いと仮定して進めます。試料をはかりで計量していますので,これはシグナルに比べキログラム原器により高いトレーサビリティを持っているからです。
この仮想直線がどの測定点にも最も近くなるようaとbを決めればよいので,ある測定点xi,yiについて考えると仮想直線上のy点とyi点の差,すなわちyi-axi-bがありますから,その二乗を全部の測定点について合計します。二乗にするのは分散と同様,正負のばらつきのある数値群の処理法です。数式で表せば,
S = Σ(yi-axi-b)2
を最小にするa, b を求めることになりますが,最小二乗法の計算手順に従えば次のようになります。
a = Σ(xi-x0)(yi-y0)/Σ(xi-x0)2
b = y0-ax0
ただしx0, y0はそれぞれ全データの平均値を表します。CHN自動分析計ではこの計算をコンピュータで一瞬に行い,検出感度や空試験値として表示します。
成分量とシグナルが単純な比例関係を持っているときは,x軸,y軸に等間隔目盛りを考えればよいのですが,なかには対数関係のものもあります。電極電位や光の透過度がその例ですが,現在では電子回路で対数に変換する技術が進み,直線で回帰するのが普通になっています。また直線回帰よりも曲線回帰のほうが測定点に合うときもあり,このときは二次曲線などを適用したりします。ただし回帰曲線は研究段階で取りあえず使うこともありますが,実用分析では曲線の癖を電子回路で補正し,直線化したものを用いたほうが便利です。
検量線からのデータのばらつきを相関係数correlation coefficient,ρで表すことも出来ます。これは本来無秩序に見えるデータからある方向に規則性を見つけ出すために考えられた手法ですが,例えば一日に吸うタバコの本数と肺がんの罹患率の因果関係などの統計があります。データポイントが一直線に並ぶときはρ=1ですが,データが円形に散らばっていて,どんな方向にも決まった線が引けないと言うときはρ=0になります。(図4)
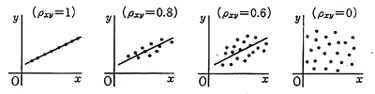
図4.相関係数
相関係数の求め方は,先ずデータ (xi,yi)がn個あるとして,平均値x0とy0からの偏差の積として共分散Cxyを計算します。
Cxy=Σ(xi-x0)(yi-y0)/(n -1)
これをxとyの標準偏差{Σ(xi-x0)2/(n -1)}1/2および{Σ(yi-y0)2/(n -1)}1/2
で割れば相関係数ρが次のように求められます。
ρ=Σ(xi-x0)(yi-y0)/{Σ(xi-x0)2×Σ(yi-y0)2}1/2
例えば表2の硫酸ヒドラジンのバリウム滴定において10),硫黄の量mgと0.005 mol/l過塩素酸バリウムの滴定値mlから相関係数を求めるとρ=0.99998となり,殆ど一直線に乗ることが分かります。CHN計の成分量とシグナルの関係も似たようなものになります。ただしこの方法では9がいくら続けば元素分析の要求精度に合うのか馴れないとすぐには分かりかねます。相関係数を求めることは数理的には間違っていませんが,元素分析の精度評価に適用するには向いていないように思います。
表2.硫酸ヒドラジンのバリウム滴定
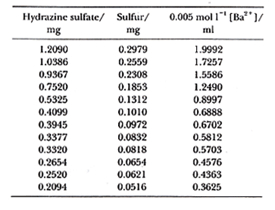
代わりの方法として検量線(回帰直線)からの測定値の偏差(成分量は正しいものとします)を統計する方法があります10)。まずそれぞれの偏差の二乗を合計し,平方根を求めます.標準偏差と同じ手法ですが,成分量全域について測定値のばらつきが分かります。表1の例で最小二乗法による検量線を求めると,
y = 6.641x + 0.0237
となりますから,検量線からの測定値のばらつきを偏差平方和として計算すると,
Σ(yi-y0)2 = 153.08×10-6
従って不偏分散平方根は,
σ= [153.08×10-6/(12-2)]1/2 = 0.0039 (ml)
ここで自由度を12-2としたのはデータ数12個のうち切片と勾配の2項目を決めたので2を減じたものです1-p135)。
滴定液1 mlは0.160 mgの硫黄に相当するので,0.0039 mlのばらつきは硫黄の量として0.000625 mgに相当します。試料量を1 mg取ったとき硫黄の分析誤差はσ=0.0625%で,2σの幅に広げると0.124%となり,これはかなり良い滴定精度と考えられます。ただしこれは無機標準を用いた滴定精度だけの評価で,有機試料では燃焼,吸収など他の誤差要因がこれに加わって誤差も大きくなりますが,検量線からのデータのばらつきを知るには分かりやすい方法ではないかと思います。
6-1.おわりに
分析値の誤差は与えられて始めて分かるもので,確率という抽象的なルールに乗っているものの,予め知ることができません。しかし統計理論の進歩でありうる範囲や,これ以上は滅多にない限界を知ることはできます。抽象的な議論ですから,言葉の定義を決めて置かなければ先へ進めませんので,誤差のばらつきや真の値からのずれなどを,標準偏差やかたよりなどと称し,これに数理的な意味づけをして来ました。
統計理論はもともと工業生産の品質管理で大きく発展したもので,分析化学はその成果を受け入れたものですから,JISなどの動きに連動します。用語などもJISが変われば一緒に変わるので,われわれには少々迷惑なことがあります。本稿には精度と真度,それに精確さや不確かさなど新しい用語の定義を説明していますが,これが定着するまでには多少時間がかかりそうです。用語がどう変わろうと,分析誤差の実態は変わりませんので,誤差を理論値との単純な比較として見ず,統計量の一部として理解するよう心がけることが大切です。
7-1.参考文献
1) 小島次雄: 分析化学における推計学(基礎分析化学講座 5),共立出版(1965).
2) 穂積啓一郎: 島津科学機械ニユース,26, No.6, p1, 島津製作所 (1985).
3) Al Steyermark: Quantitative Organic Microanalysis, p7, Academic Press, (1961).
4) C. Rodden, et al: Ind. Eng. Chem. Anal. Ed., 15, 415 (1943).
5) 元素分析懇談会編: 最近の元素分析法(化学の領域増刊 19),p14,南江堂 (1955).
6) 有機微量分析研究懇談会編: 有機微量定量分析,p13, 南江堂 (1969).
7) 清水正郎,穂積啓一郎: 分析化学,19, 1041 (1970).
8) 日本工業規格: JIS Z8402-1 (ISO 5725-1) 日本規格協会 (1999).
9) 内川恵三郎,穂積啓一郎: 分析化学,21, 232 (1972).
10) 穂積啓一郎,佐藤綾子,秋元直茂: 分析化学,47, 219 (1998).