



TOPICS
CHNフォーラム
第四部 物質の純度と精製
1-1.はじめに
混じりけのない物質を「純粋」と言いますが,これは「真空」と同じように概念としてはあっても実際には得難いものです.物質は無数の分子や原子から出来ていますが,1モルの物質は6×1023個の粒子の集団ですから(アボガドロ数),例えばアセトアニリド1mgはC8H9NO=135.16から計算して,
6×1023×(1×10-3/135.16) =6×1020/135.16 =4.4×1018
と言う数(4.4兆の百万倍)の分子になります。耳掻き一杯にも満たない物質量ですが,驚くべき数の集団です。これに比べると地球上の人口の50億は5×109人になり,上の数より10桁ほど少ない数値になりますが,それでも結構いろいろな人種があってお互い異文化の衝突があり,人類は中々均質になりません.純粋とは構成要素がすべて同じ規格にはまらないといけませんが,アセトアニリド1 mg中の分子(4.4×1018個)がすべて同じ化学構造のものであることは現実には無理な話で,多かれ少なかれ異物が含まれています.似たようなことは真空の定義も同じで,ガラス球をポンプで排気して殆ど気体のない空間にすることはできますが,そこは圧力が極めて低いというだけで,気体分子がゼロと言う空間にはなりません。地球上が無理でも宇宙空間に出れば真空があると思われますが,そこでも星間ガスが僅かに漂っていて,本当の真空は存在しません。
概念と現実が一致しないことは昔から経験してきたことで,それ故に概念に向けて現実の改善を図ろうとする原動力が生まれました。砂糖黍から甘味の本体を取り出すために,絞り汁を煮詰めて黒砂糖を作る技術は古くから沖縄で進歩し,江戸時代薩摩藩が独占してドル箱になりましたが,その後徳島藩で精製度の高い三盆白が出来てもっと上品な味の高級和菓子が作れるようになりました。甘味成分の繰り返し結晶化による不純物の除去がこれを可能としました。現在はもう通用しませんが,かつては黒砂糖で作ったものを駄菓子,三盆白で作ったものを上菓子という区別がありました。食物に欠かせない塩も同じで,岩塩の取れないわが国では海水が主な原料になりましたが,古代はホンダワラなど海草を乾燥して焼く藻塩から始まり,のち海水を汲み上げて煮詰めるのが主となりました。瀬戸内海は日照りがよく,塩分も濃いので砂浜は大規模の塩田となり,赤穂藩はじめ塩の産地として栄えました。塩釜で煮詰めて水分を蒸発させ,塩を結晶に出して集めるので,かなり効率のよい精製法でしたが,塩化カルシウムや塩化マグネシウムなど「にがり」が取り切れず,吸湿性でべたべたした塩ができました。赤穂の塩が良質で高値で取引されたのも,にがり除去の技術が優れていたからと言われます。
再結晶(recrystallization)はもともと固体を溶媒に溶かし,この溶液から溶媒を蒸発させ,溶け込んでいた物質を再び固体結晶として析出または沈殿させることですが,結晶の成長のとき溶液中で勝手な方向に動いていた成分分子が,一定の秩序ある積層として固体に成長し,液相から分離します。溶液から溶媒が蒸発して溶質(成分)が濃縮されると溶質どうしの接触が増え,もし相互に分子間力が働くと結合したまま動かなくなることも多くなります.有機化合物では一般的にファンデルワールス力(van der Waals force)という,分子の内部で電子が動き回っているために生じる電子密度の偏りから分子の極性または電気双極子が生まれ,その他にも水素結合など原子間の結合力が働いて相互に引き合います。同じことが繰り返されると溶質分子は溶媒を排除しながら積層し,結晶化します。このとき溶質分子は最も安定なエネルギー状態を求めて図1のように細密充填の形に並びます1)。細密充填では(a)の平面層から2層,3層と隙間無く溶質分子が並ぶので,溶媒分子や異物の入り込む隙間がありません.理想的にこの細密充填が進めば一度の再結晶で高純度の溶質結晶が得られるのですが,なにしろ耳掻き一杯のアセトアニリドには4兆の百万倍もの分子があるのですから,結晶成長の過程であちらこちらに主成分と違う不純物分子を取り込んでしまうのは仕方がありません。 出来た結晶を濾過や遠心分離機で分離してもう一度溶媒に溶かし,再結晶を繰り返せばまた純度は上がりますが,だんだん主成分が無くなって来て少量,微量の物質では何回も再結晶の操作を繰り返すことが出来ません。またあまり純度が上がると今度は溶媒中の不純物や容器からの汚染が問題となってきて,ここでも限度があります。

図1 分子の細密充填構造
定量分析では標準物質や標準試料が重要なよりどころとなっていますが,どちらも公的機関や学会で認証された規格値を持っています。多くの場合純物質を用いますが,この他にビタミンやホルモンなど生物活性を規制するものや混合ガスの濃度を保証するものもあり,そのときは純物質でなくとも力価や濃度を保証して標準品とか標準ガスなどと呼ぶこともあります。有機元素分析では当然純物質を標準試料としますが,それは純物質において1分子の化学構造から計算される元素の含有率が,物質全体の元素の含有率と一致するからです。現実的には標準試料に僅かな不純物があっても殆どこの一致に影響を与えないことで純物質並の扱いがされています。このため日本分析化学会有機微量分析研究懇談会では標準試料検定小委員会というボランティアグループを作って,定期的に標準試料の品質管理を続けています2)。試薬メーカーから提供される殆どの標準試料が規制に合格しますが,時には不合格品が見つかり,試薬メーカーに判定結果を伝えて,標準試料の精製過程や保存管理に問題がなかったかの検討を要請しています。
標準試料が定量分析の重要なよりどころとなったのは時代の推移が大きく関っています。初期の定量分析では重量法,容量法が主体で,データの計算は試料の質量,生成物の質量,滴定液の容量などの数値で行われました。滴定液の容量もビュレットの目盛りを水の質量で検定しますから,すべての定量分析の基準が質量に回帰し,この中でデータ処理が完成しました。極言すれば標準試料など無くてもよかったことになります.それにも拘わらず標準試料はアセトアニリドなど元素分析の初期から使われていて,目的は目下の分析手順が正しく行われて,燃焼や吸収,沈殿反応などが定量的に進んでいるか,そして最後に試料の理論含有率を正確に言い当てるかどうかの確認に用いられました。
大きな転機がやってきたのは1960年代の分析機器の電子化です。分析反応で得られる生成物の量や濃度を光や熱などのセンサーで拾い上げ,得られた電気信号を増幅,記録させるので,分析方法も一変しましたが,得られた電気シグナルを質量に変換しなければなりません。センサーに届いているはずの生成物量を言い当てるために,どうしても成分量既知の標準試料が必要となりました。標準試料であれば秤取った量から生成物の量も計算できます。生成物の質量と電気シグナルを対応させることで,シグナルは生成物の質量に換算できるようになりました。こうなると分析データの信頼性は標準試料の元素含有率に丸々依存することになり,標準試料が無くては分析が出来ないという現状を生んでいます。
標準試料はわが国ではキシダ化学(株)によって50種近い化合物が提供されています3)。元素構成ではC, H, N, O以外にハロゲン,硫黄などを含むものがありますが,一方化学構造上の特徴として環状や鎖状構造のものや高分子化合物も選ばれています。殆どが固体結晶ですが,中には液体試料もあり,サンプリングに手際を要するものがあります。いろいろな性格を持った標準試料が選べるのはよいことですが,反面試料物質の種類が多く製造面や保存の面で技術的な問題も残されています。本稿では標準試料に関連して物質の精製法や純度の鑑定に関する基本的な事項を解説します。
2-1.再結晶による精製
溶液中の成分を結晶として取り出す再結晶法が,物質の精製に効果的であることは上に述べましたが,成分によって結晶の出やすさや形態はそれぞれ違っています。成分分子の立体構造が四角いもの,丸いもの,平たいもの,細長いものなどがあり,従って積み上がった結晶の形も違ってきます。また総ての分子が決まった方向に向いているので共通軸を持っており,光を当てたり,圧力を掛けたりすると方向によって違った性質や応答を現します。この「異方性」というのが結晶の特徴で,これに反し分子の並び方に規則性のないガラスや自由運動をしている液体,気体は,全体としてどこから見ても同じなので「等方性」を持っています。異方性か等方性かを見分けるには2枚の偏向板で挟み,一枚を回転して物体が暗くなったり明るくなったりするかどうかで分かりますが,正確なことはX線回折像によって判断できます。規則性のある層状構造を持った固体にX線のスポットを斜めに当てると反射光に同心円の回折像が現れます。
X線は原子や分子の間隔に近い波長を持っているので,反射光は位相が合って強めあう方向と弱めあう方向があり,検出器でX線強度を角度を変えて記録すると所々に強めあう方向のピークが現れます。図2はシユウ酸カルシウムのX線回折像ですが,検出器の角度2θを大きくすると左から右に決まった位置に鋭いピークが見られます。これに反し結晶性でないものは雑音信号しか現れません。結晶性物質は少々砕いて粉末にしても,X線の波長よりは遥かに大きいので,X線回折像にあまり変化はなく,通常測定は粉末試料を用いています。

図2 X線回析装置と回析増
結晶は分子間力で相互に規則正しく積層したものですが,結合の強さはいろいろです。無機物はイオン結合が多いので+-の静電力で強力に引き合い,少々熱を加えても結合が緩まず一般的に融点が高くなります。これに反し有機物はファンデルワールス力や水素結合など弱い力で結ばれているので,少し温度を上げるとこれらの結合が外れ,融解したり昇華したりします。融解も昇華も分子が自由に動きだすことが原因ですが,要するに結合エネルギーに等しい熱エネルギーを加えると起こる現象です。非常に純粋な物質ですとどの分子も同じ結合力ですから,ある温度で一斉に自由になり,鋭く再現性のある融点が得られます。これに比べると不純物を含むものは結合力があちこちで阻害され不ぞろいになりますから,純物質より低い温度で融解を始めます。融点降下と呼んでいますが,溶け方もある温度で一斉には行かなくて,温度幅を持って溶けて行きます。
融点は物理化学的な定数で,固体と液体の相変換を行う温度ですが,圧力にも多少影響されます。水の三重点というのが状態図(図3)によく例示されますが,ここでは三重点は0.01℃で,1気圧のとき0℃で氷と水が平衡状態になります。このまま熱を加えてゆくと氷は融けてゆきますが温度は不変で,最後に氷が無くなると水の温度が上昇します。100℃に達すると水の蒸気圧が1気圧になり,沸騰をはじめます.実験をゆっくり行えば氷の融点は精密に測定できます。どの固体物質にも同じような状態図が描け,これから融点も測定できますが,この方法は相当量の試料を必要とし,また時間もかかりますので,通常の化学実験には間に合いません。特に微量物を対象とすることが多い有機化学では,手軽で迅速な融点測定法が求められ,このため数ミリグラム以下の試料で行う微量融点測定が通例になっています。
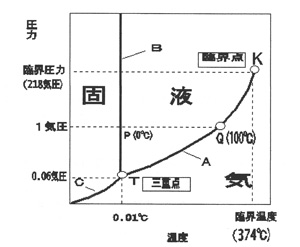
図3 水の三重点
融点の観察は古くから物質の同定や純度の判定に使われましたが,目視で行うものから,もっと精密に解析するために少々複雑な熱分析の装置がいろいろ開発されました。最初は試料を粉末にしてガラス毛細管に詰め,温度計を入れた硫酸浴の中でゆっくりとガスバーナーの炎で温度を上げてゆく融点測定器が作られ,長い間利用されました4)。大抵の有機化学実験室には実験台ごとにこの道具が置いてありましたが,硫酸浴をうっかり壊すと熱硫酸が机上に流れて木を焦がしたり火傷をしたりすることがありました。戦後になってシリコーン油が出回るようになって薬局方試験法5)やJIS規格6)など公定法に取り入れられるようになり,硫酸浴の危険は無くなりましたが,測定原理はそのままで,現在もこれら公定法で実施されています。器械装置が要らず,ガラス器具だけで済むので法制面で統一し易いからでしょう。
硫酸浴を用いる融点測定管は図4-aのように温度計をコルク栓に通して固定し,試料を入れたガラス毛細管を温度計水銀部に接触させ,下からマイクロバーナーで加熱しました。当時は温度計が全没式であったので,正確な融点測定にはbのようにもう一本の細い温度計をゴム輪で取り付け,露出部の温度を知り,次のような補正式によって浴温すなわち融点温度を加算補正しました。
補正温度(℃)=浴液から上の温度目盛×(浴温度の読み-露出部温度の読み)×0.00016

図4 融点測定管
測定値に毎回補正値を計算して加えると言うのは煩わしい作業なので,日本薬局方第8改正では図5のような浸線付き温度計に改め,加熱浴のシリコーン油のメニスカスを浸線に合わせると浴温がそのまま読み取れるようにしました。温度計も50℃レンジの6本組みで見やすくなるとともに,加熱浴の構造も多少進歩して使い易くなっています。この構造はその後JISにも採用され,現在に至っています。

図5 日局融点測定器
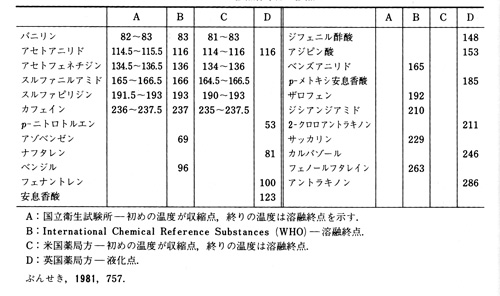
純物質はある温度で一斉に融けるのが原則ですが,実際には試料粉末の詰め方や温度の上昇速度に不均一があったりして,文字どうり一点では融解しません。加熱浴の中の毛細管を観察すると図6のように融点付近で一連の変化が現れ,どこを融点とするかは制度や国によて違っています。国際的には融点を融解範囲(melting range)で示すことが多く,わが国のJISもこれを採用していますが,この方法では収縮点から融解終点までの温度を取っています。しかし融点の認定法は国によって多少バラエティがあり,米国薬局方と国際薬局方では融解範囲を採用していますが,英国薬局方では液化点を,日本薬局方では融解終点を読み,その読みが規定の範囲にあればよいということにしており,融解範囲と紛らわしい表示になっていますので注意が必要です。取り決めによって認定された融点標準品が各国にありますが,それらを纏めた例を表1に示しました。

図6 融点付近の外形変化
表1 各国標準品の融点
3-1.固体物質の精製
純物質に不純物が混入すると通常融点が下がりますが,純度の判定によく使われます。希に融点に変化が無かったり,また逆に融点が上がったりする場合もありますが,滅多にない特殊なケースです。純物質に混在する不純物は一種類と限りませんが,仮に一種類とすると二相系の混合物となります。純物質Aに不純物Bのモル濃度が増えると融点は下がり,さらにBが増えるとA:Bのあるモル比で最低の融点を観測しますが,これを共融混合物と言っています。さらにBの濃度が増えると再び融点が上がりますが,最後にB成分の融点に達します。図7はこのような二相系の温度―組成図ですが,e1は共融点のモル組成,aの縦線はある組成での温度による状態変化,bはある温度における固体の範囲を示しています.AとBの種類が決まっていて状態図が与えられていれば,不純物Bの存在量と融点降下は多少数値的に結びつけられますが,普通不純物の種類や濃度は分かりませんから,要するに融点が純物質より低ければ不純物が含まれていると判断します。逆に再結晶で精製しても融点が上がらなければ,これは単一成分の純物質と考えてよいでしょう。
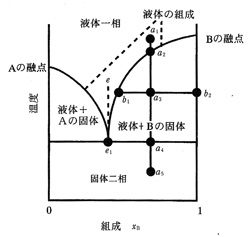
図7 二相系の温度―組成図
標準試料を製造する試薬メーカーは専門的な精製技術を駆使して純物質を提供していますが,物質によって精製し難いものや,製造後変質するものがあり,いろいろ苦労しています。多くの場合化学工場で生産された物質を購入してさらに精製しますが,当然固体物質は再結晶法により純度を上げます。再結晶法は標準試料の製造に限らず,一般合成化学や天然物の成分研究にも繁用されますので化学操作の重要なプロセスです。原則的には固体物質に有機溶媒を加え,温めて溶かし,冷却すれば目的成分の飽和液の中に結晶が析出して沈殿します。しかし飽和溶液に溶けたまま結晶にならない分は,濾過しても回収できませんので損失となります。結晶の純度を上げることと,回収率をよくすることは二律背反になりますので,このあたり化学者の判断が必要です。
再結晶溶媒の選択に当たっては,物質によって溶媒への溶解性の高いものや低いものがあり,出来れば不純物の溶解性が高く,かつ目的物の溶解性がこれより低い溶媒条件が望ましいのですが,そう都合のよい溶媒は見つからないので,水と混ざる有機溶媒を選び,混合比を変えて溶解性を調節します。水―メタノールがよく使われる例ですが,混合比は試行錯誤で探さなければなりません。固体物質に溶媒を加えて温めて溶かし,一度濾過してビーカーに貯め,室温に放置すれば結晶が析出します。結晶は出やすいものや出にくいものがありますが,出にくい時はガラス棒でビーカーの底を擦るとか,原料固体の極微量を種として加えるとかで結晶化を促進することが出来ます。冷蔵庫で冷やすのも方法ですが,不純物も一緒に沈殿しやすいので注意が必要です。
溶液を濾過したり,出てきた結晶を分離するために濾過用器具がいろいろ工夫されてきました(図8)。濾紙は繊維の網目を利用して固相と液相を分けますが,どうしても濾紙の細かい繊維屑が濾液に流れ出てきて,このあと精製の邪魔になります。(e)のガラスフィルターはこの心配がないので高度の精製には向いています。目の細かさが表2のように1号から4号まであって番号順に細かくなっていますが,3号あたりがよく使われます。よほど細かい沈殿を濾過するには4号を用いますが,濾過に時間がかかり,吸引濾過が必要です。
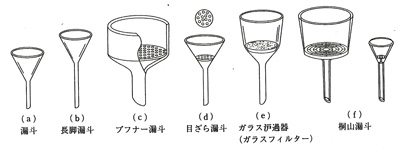
図8 各種濾過装置
表2 ガラス濾過管の目の規格

ガラスフィルターは化学実験の必需品でどこの実験室でも机の引き出しにゴロゴロしていますが,何時の時代に誰が考案したのかはっきりしませんでした.これについては本解説記事「13.超微量分析の技法」の中で少し触れてありますが,たまたま金沢大学医学部の岩崎憲名誉教授がドイツ留学中,オーストリアのプレーグル研究所の微量分析技術コースで研修を受けられた話を京都大学でされました。そこではベルリン滞在中生体成分のリンの定量にケルダール分解を行い,リン酸をモリブデン試薬で沈殿させ,フィルターで沈殿を濾過して重量を計る方法をとっていました。この沈殿は極めて細かく,濾紙の目を通り抜けてしまうので,当時は目の細かい沈殿はアスベストをガラス管に詰め,静かに濾過するのが通例でしたが,アスベストの繊維が僅かに流れ出して定量の障害になりました。
岩崎教授はアスベストが流れ出さないようにガラス粉末を板状に焼結し,ガラス管に溶接するようエナ市のショットガラス社(Schott AG)に依頼されました。こうするとアスベストは流れ出ないことと,広い濾過面積で効率よく分離ができ,ベルリンの研究所で重宝されました。プレーグル研究所の研修を受けに行くときこのガラスフィルターを持参して所長のプレーグル教授に見せたところ,時間を掛けて見た上,「これはうまく出来ているので,君が実験の時使いなさい,ただし私は今までの濾過法でよい」と改めなかったと言います.少々頑固な人であったようで,自分は左ぎっちょであったせいか,横型の燃焼管にキャリヤーガスを左から右に流し,試料の入り口も左側にするなど,右利きの多い弟子達も迷惑したかと思いますが,存命中は押し通しました。
1930年プレーグル教授が亡くなってロート教授(H. Roth)が所長を引き継ぎ,その後1935年には微量分析のテキストの改訂版がでましたが,早速燃焼管のキャリヤーガスは右から左向きに変更され扱い易くなりました。驚いたことにこの改訂版には岩崎教授考案のガラスフィルターが掲載されていて,簡単にショット社の製品とあり,これを見た岩崎教授はびっくりされたと言うことです。故意というよりプレーグル教授が事情をロート教授に詳しく伝えなかったため,考案者が誰か分からず製造社の名前だけが残ったのでしょう。最近発明者と製造会社の間で対価のやり取りが話題になっていますが,世界で使われるガラスフィルターの数量は膨大なもので,発明者のことはもう済んだ話ですがせめて日本人だけでも岩崎教授の発明の功績を記憶に残したいものです。
話が少し外れましたが,綺麗な結晶を得るにためいろいろ工夫が必要です。結晶の成長が溶媒を排除しながら行われることから,理屈の上で大きな結晶を成長させるのがよいように思われますが,これは少し問題があります。結晶成長過程で例えば洞窟のようなものが出来て溶液を取り込んでしまうと,あとで減圧乾燥しても出てきません。水晶の結晶中に古代の水が閉じこまれ,これが装飾品になったりしていますが,これと同じ現象です。どうしても大きな結晶になってしまう時は乳鉢である程度つぶして粒度を揃え,減圧乾燥する必要があります.標準試料を作るような時は原料も相当きれいなものですが,一般の合成化学や天然物化学の研究過程では褐色の着色物質が混じることがよくあります。単に着色物を除去する目的であれば,活性炭を溶液に加え着色物を吸着させ,濾過すればかなり除去されますが,ごく僅かな炭素がコロイド状になって濾過材を通り抜け,その後の元素分析で炭素値を高くすることがあります。元素分析の前段階では活性炭による脱色は避けなければなりません。再結晶で色が多少残っても活性炭処理よりは安全です。
再結晶法は精製の王道ですが,近年扱う物質量が少なかったり,手っ取り早い精製法として液体クロマトグラフィがよく使われるようになりました。ここではカラムから出る分画成分を取り分け,溶離液を蒸発させた残分を精製品としていますが,再結晶品とは違った問題が残っています。クロマトそのものは強力な分離法ですが,取り出した場所で本当に単成分だけになっているかどうか分かりません。化学特性が似た不純物と重なっている可能性があります。溶離液を減圧で気化させますが,再結晶の時に溶媒分子を排除する力と同じくらいの気化力がないと同じように除去できません。成分と溶離液の間の分子間力は相当に強いので溶けているわけですから,これを引き離す力が要ります。もう一つの問題はクロマトカラムの充填物のかけらやコーティング材が流れ出て,その微粒子が混在することになります。カラムの末端に多孔性のプラグが入れてありますが,これより小さい浮遊粒子は通り抜けます。一般にクロマトで精製した物質は結晶体にならず飴状のものが多く,外見でも結構見分けがつきます。化学者も対象物の量が少ないので,止むを得ずクロマト法を使うことになり,この点理解できますが,これを依頼分析の試料として持ち込まれる方はもっと難儀します。この試料は多分目標物質でしょうから分析値は計算値に近いものが得られますが,少し計算値から外れるので試料が悪いのかそれとも分析の誤りか判断を迷わせます。このような傾向は当分続くと思われますが,最近は手近に質量分析や核磁気スペクトル装置がありますから,これらのデータと照合すれば目的物かどうかの判定ほぼ確実となります。
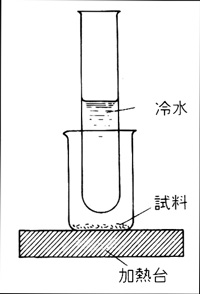
図9 試験管による昇華法
混合物のうちある成分が熱で気化しやすい時は昇華法で分離できます。気体の蒸気から冷えたガラス面に結晶化するので,精製法としても用いられます。数mgの物質でも応用可能ですから,微量分析の技術として優れています。試験管を用いる昇華法では図9のように小ビーカーに粉末の原料物質を入れ,上から冷水を入れた試験管をスタンドで保持し,ビーカーの下から加熱台で熱します。気化した成分は試験管の外側に結晶化しますから,あとでスパチュラを用い掻き落します。もっと少量の物質では図10のように二枚の時計皿を貝殻状に合わせ,中に物質を入れ,下から加熱して上の皿に結晶を作らせることも可能です8)。
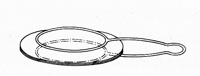
図10 時計皿による昇華法
昇華物質を純粋に得るには熱分解を避けるためあまり高い温度をかけないことが必要です。減圧下の昇華がこの点安全です。図11はその例で,物質は外管の底に入れ,冷却ジャケットには水,氷,ドライアイスなどを入れて冷やし,ポンプで減圧しながら,器具の下から加熱して昇華させます。
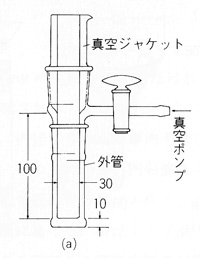
図11 減圧昇華装置
4-1.液体物質の精製
沸点の違う2成分の混合物は蒸留によって分別することが出来ますが,ワインやビールからアルコールの濃厚なブランデーやウイスキーを造るプロセスと同じです。図12はヘキサンとヘプタンの気液組成ですが,双方50%の混合液を80℃近くにすると気相のヘプタンのモル分率は30%ほどに下がり,代わりにヘキサンのモル分率が70%ほどに上昇します。これを冷やし液化してもう一度温めると更に気相のヘキサンのモル分率は90%ほどになります。ステップを増やせば気相のヘキサンモル分率はどんどん上がりますが,この繰り返しを小刻みに行う器具が分別蒸留装置で,例えば図13のようにビグローカラム(Vigreux column)8)と称する蛇管を数mlほど液の入ったフラスコの上に取り付け,下から加熱すると沸点の低い成分が気相と液相の平衡を保ちながら上昇し,最後に受器のほうに流出します.図は減圧下の操作を示していますが,受器を回転すると異なる沸点の物質を取り分けることができます。特定の受器から成分を取り出せば精製の目的が達せられます。
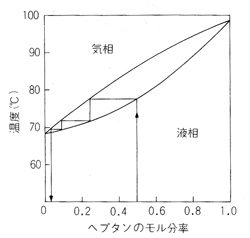
図12 ヘキサンとヘプタンの組成図
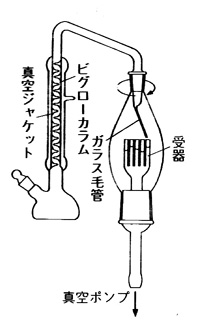
図13 ビグローカラム
1 ml前後の液の蒸留では図14のような簡単な器具が使えます。丸い蒸留フラスコの上にガラス管を取り付け,吹きガラスの方法でフラスコをガラス管に押し込んで環状のひだを作ります。留液はこのひだに溜まるようになっています。温度計を挿入して蒸留温度を知ると同時に蒸気の還流をさせます。フラスコにはガラスウールを入れて多少精留効果を与えます。フラスコを下から加熱すると沸点の低い成分から気化し,ひだの部分に集まりますから,長い毛管ピペットで吸出します。最初の成分より2番目の成分が必要な時は,最初の成分に溶媒を加えて洗い,温度を上げて2番目の成分を蒸留します。この器具は常圧でも減圧でも利用できます8)。
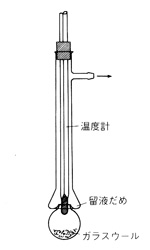
図14 微量試料の蒸留
手軽な道具を紹介しましたが,目的成分と沸点が30℃以下しか違わない不純物を分別するには精留カラムが必要です。最良の場合1℃の分別ができますが,こうなると装置が次第に大きくなり,器内のホールドアップが増えて微量の物質は扱えなくなります。 数mlの物質の分留装置を図15に示しますが,長さ150 mm,内径2.5 mmの真空ジャケット管Aにステンレス鋼のコイルBを封入しています。装置全体を発熱線Cで巻いて保温し,沸点よりやや低い温度に保ちます.留出液は冷却器Dで凝縮し,突起Eから滴になって受器に落ちます。受器を回して異なる沸点の物質を集めることができます.この他にも液体物質の分離,精製法はいろいろありますが,固体物質の再結晶ほど純度の高い成分を取り出す方法はありません。
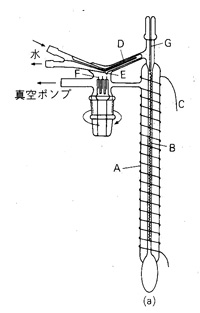
図15 精密蒸留装置
5-1.微量物質の融点測定法
融点は固体物質の固有の物性値ですから,物質の認定や純度の評価に動かし難い証拠になりますが,測定は固相と液相の熱平衡の下で得られるので,物質量もかなり必要になりますし,時間もかかります。永久不変の物理化学データを決定するには仕方がありませんが,先を急ぐ実験化学者には短い時間に融点を測定しなければなりません。また天然物化学や生化学の研究が進むと扱う物資が少量,微量になり,mgからμgの量で測定せざるを得ないことになります。毛細管に詰めてシリコーン油で加熱する公定法も割合微量で済みますが,これ以下では顕微鏡で結晶片を観察しながら融点を測定しなければなりません。この方法では結晶片個々の融解が見られるので,場合によっては二種類の物質の融解状況が同時に調べられます。
1930年代からオーストリア,インスブルック大学のコッフラー教授夫妻 (L.Kofler, A. Kofler)による一連のホットプレートと顕微鏡を組み合わせた融点測定技術が展開されましたが,結晶の光学的解析まで含まれた大きな研究成果でした。米国のアーサートーマス社がこのモデルをコッフラー微量融点測定器として製品化し(図16),広く世界中に普及しました。熱容量のある金属円板の中央に孔を穿ち,金属ドラムに収容して顕微鏡台に載せます。試料はスライドガラスに載せ,中央の穴の上に置き,さらにドラムをガラス円板で覆って電熱線でゆっくり加熱します。金属円板は温度計を差し込んで顕微鏡を見ながら融解温度を記録します。色々な小道具も付属していて,測定後温度を速やかに下げるための冷却ブロックや昇華装置,その上融点標準試料まで準備してあります。
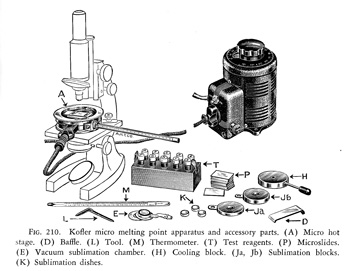
図16 コッフラー融点測定器
1950年ごろ始まったわが国の微量分析研究の分野に微量融点測定法の改良が含まれました。コッフラーの装置は加熱台の熱容量が大きく,融点付近でゆっくり温度が上昇するのでよいのですが,融点まで温度を上げるのに時間がかかり,また途中で温度を調節しようとしてもすぐには応じてくれません。熱容量を小さくして応答を速め,融点付近で電力の微調整をするのがよいと結論されました。また試料の融解と温度計の読みを目を離さず行う方式が考案されました。図17に外観を示しますが,30×17cmの上面を持つ金属ケースに加熱台,温度計,電源スイッチを取り付け,温度計と平行にスライドできるプリズムを配置しています。試料の観察は加熱台中央の小孔で行いますが,10倍のルーペか60倍の顕微鏡が選べるようになっていて,一方の目で試料を見ながらプリズムを滑動させると,温度計の目盛りが片方の目でファインダー内に写ります。プリズムには拡大レンズが組み合わせられ,虚像の明視距離25 cmを保っていますから,プリズムの位置に拘わらず温度目盛を正確に読むことが出来ます。
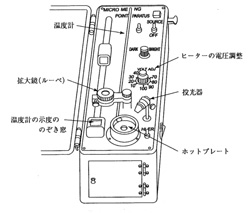
図17 微量融点測定装置
温度計はアルミニウム製の加熱台に挿入しますが,浸線付きのもので露出部の温度補正は必要ありません。30~270℃を測定範囲としていますが,一時的には300℃まで測れます。加熱台の下にはランプハウスがあり,必要により偏光フィルターを挿入できます。顕微鏡のアイピースに同じく偏光フィルターを入れると簡易な偏光顕微鏡となり,結晶体を暗視野に観察できるようになります。加熱台の温度は100 V範囲の粗調整スイッチと,これに10 Vを加える微調整スイッチで行いますが,融点未知試料の測定の時は粗調整スイッチで大体の融点の見当をつけ,このあと微調整スイッチを併用して精密に融点を測ります。試料は薄手のスライドガラスを1/3に切り,微量の試料を散布し,この上にシリコーングリースを付着させたカバーガラスを貼り付け,加熱台の中央に置きます。蓋ガラスを被せランプを点灯して測定を開始します。
温度計には補正表がついていますので,正確を期するには測定値を修正しなければなりません。しかしこの他にも試料物質と測定温度との間に誤差要因が含まれていて,加熱台から温度計水銀部へ熱伝達の時間的遅れ,加熱台からスライドガラスを透る伝熱遅れ,カバーガラスからの放熱,また加熱台中央の小孔は伝熱が弱められ,温度が僅か低下します。加熱台の温度上昇を速くするとこれらの誤差は増えるので,経験上融点付近は1分2℃くらいの上昇がよいように思われます。いずれにしても多少の誤差はあるので,正確な融点を求めるには予め決められた方法で融点標準物質を測り,標準値との差を記録して未知試料の融点を補正しなければなりません。わが国では国立衛生試験所が保証した融点の標準品を提供しており,自分の使っている融点測定器の校正をすることが出来ます。
6-1.熱解析法
融点は物質の融解現象の一点を捉えていて,簡単な道具で測定可能ですが,融解前後の物質の熱的挙動までは知ることが出来ません。物質の温度を室温から徐々に上げて行くと途中で結晶構造が変化(相転移)したり,融解,昇華,熱分解,脱水などの現象が起こることがあります。多くの場合物質が他から熱エネルギーを吸収するのでその現象が終わるまで温度が上がりませんが,希に発熱してその現象が終わるまで温度が戻らぬ場合もあります。測定物質が微量になるとこの熱量も小さいので鋭敏なセンサーが必要です.方法としては加熱によって熱変化を受けない基準物質,例えば酸化アルミニウム(α-Al2O3)と試料物質を並べて加熱炉に入れ,ゆっくり温度を上昇します。それぞれの温度を熱電対によって検知しながら記録計に描くと,基準物質は直線的な温度上昇を示しますが,試料物質はところどころで温度遅れや停滞をします。それぞれの温度が何度であるかより,基準物質と試料物質の差を読み取るほうが精密に行えるので,示差熱分析(Differential Thermal Analysis,DTA)の装置が作られました9),10)。
図18に示差熱分析装置の概略図を示しますが,温度差は二本の熱電対を逆向きに繋いで取り出すことが出来ます。基準物質の温度をベースラインとして記録紙上に描かせると温度差の時間的変化を示差熱分析曲線として得ることが出来ます。ベースラインに対して吸熱変化(融解など)は下方に,発熱変化(酸化など)は上方にピークを生じます。融点で生じる吸熱ピークの形を標準試料と比較することが出来,ピークの立ち上がりや広がりである程度純度を評価できます。
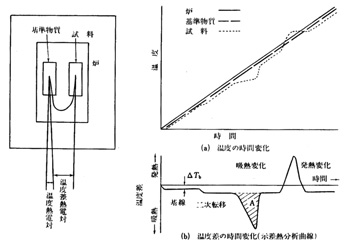
図18 示差熱分析装置の概略図
その後もっと精密な熱解析に示差走査熱量計(Differential Scanning Calorimeter, DSC)が開発されました9),10)。構造的にはDTAと似ていますが,基準物質と試料物質との温度差を測るのではなく,例えば試料物質に吸熱変化があるとき,微弱な電力を加えて基準物質と同じ温度に保つ方法がとられています.電力補償方式とも呼ばれていますが,この電力は吸熱する物質量に対応していますので,定量性もあり,微量分析には利用価値の高い方法です。図19に原理図を示しますが,基準物質と試料物質の容器に何れも共通ヒーターと差動ヒーターを取り付け,共通ヒーターには温度プログラムに従って電力を少しづつ供給します。基準物質と試料物質に温度差があると,これを感知して差動ヒーターに電力を加えます。つねに両者が同じ温度を保つ状態で供給電力を記録すると,試料物質の吸熱または発熱履歴がピークとして得られます。記録パターンはDTAと似ていますが,基準物質も試料物質も同温ですから,加熱炉の雰囲気内で熱的に安定し,測定値の信頼性も高いという長所があります。標準試料の走査パターンと比較すると,未知試料の同定や純,不純の推定が可能となります。
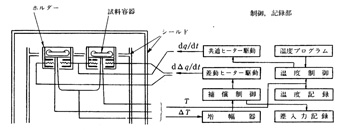
図19 示差走査熱量計の構成図
7-1.おわりに
無数の分子から構成される物質は,ランダムな発散と結合を繰り返した結果生まれるものですから,単一の種類の分子だけの集まりと言うのはは自然界にありえない現象と言えます。水晶や雨水のように比較的単純な分子の集まりは珍しいことで,殆どの元素や化合物は複雑な構成の物質から物理的,化学的な方法で取り出し,さらに単一成分になるまで精製をしなければなりません。化学工業では大量の原料から化学物質を生産し,それぞれを出来るだけ純粋に近づけて市場に提供していますが,利用目的が試薬,医薬品,農薬,標準品などとなると僅かな不純物までが問題となります。自然には存在しない純粋物質をそれに近いものとして作る技術が精製法ですが,精製しやすいものや困難なものもあり,出来上がった物質の純度は100%とは言えません。本稿では高純度を得ることの難しさを前提に,それでも精製をどうするか,またどのように精製度をチェックするかについて一般的な事項を解説しました。
精製する物質の形態は固体,液体,気体が含まれますが,実験室で精製できるのは固体が主で,液体や気体はあまり効果的ではありません。分子運動が小さい固体では分子間力がよく働いて同一分子の積層ができ易く,不純物の排除が有効に行われます。これに比べると液体や気体は分子運動が激しくて,勝手な方向に動いていますから,纏まった集団になりません。液体の場合は主として蒸留法に頼っていますが,分子量に依存する運動エネルギーの大きさで低分子種から気化する性質で分離します。しかしあまり歯切れのよい分離法ではないので固体の再結晶のようには行きません。気体も極低温で液化して蒸留法で分離できますが,実験室で簡単に出来るようなものではありません。いずれにしても固体物質には少量,微量でも再結晶法が適用できますが,液体,気体では量がないと精製は困難という宿命があります。
8-1.参考文献
1)田中勝久:固体化学,pp11,東京化学同人,(2004).
2)日本分析化学会有機微量分析研究懇談会:標準試料検定小委員会
3)有機元素分析用標準試料:キシダ化学株式会社,大阪市中央区本町橋1-22.
4)日本分析化学会編:分析化学実験ハンドブック,pp51,丸善,(1987).
5)第11改正日本薬局方解説書:融点測定法,B-381, 広川書店,(1986).
6)日本工業規格JIS K0064:化学薬品の融点及び溶融範囲測定法,日本規格協会,(1992).
7)Pregl-Roth:Quantitative organische Mikroanalyse, 2 Aufl. Springer, (1935).
8)穂積啓一郎:微量化学の手法,pp128, 157,化学同人(1981).
9)日本化学会編:実験化学講座 4, pp59, 丸善,(1992).
10)日本分析化学会編:機器分析ハンドブック,pp359,丸善,(1996).