



TOPICS
CHNフォーラム
第八部 燃焼と分解
1-1.はじめに
燃焼という現象は人類もその他の動物も有史以前からずっと見てきたものですが,副産物の熱と光を生活に利用することで人類の文明が生まれました。山火事や雷など自然界の火を待たずに,木をこすり合わせたり火打石で火花を散らせたりする人工発火法は偉大な発明で,好きなときに火が得られ,豊かな生活を保つため火は欠かせないものになりました。ギリシャ神話ではプロメテウスが天上から火を盗んで地上の人間に与えたことになっていますが,この話はそれほど貴重なものをわれわれが手にしたことを教えています。東洋では火は清める効果が強調され,燃やしてしまえばすべての罪や穢れが消えてしまうと考えました。ヒンズー教や仏教が古くから火葬をするのもそれなりに理にかなっています。
物体を燃やすと僅かな灰を残してほとんど消えてしまうことは長い間誤解を招きました。錬金術がまだ生きていた中世にはフロギストン(燃素)説というのがかなり長い期間定着しましたが,ここでは 「物体=灰+フロギストン」 で,物体に熱をかけるとフロギストンが抜け出し灰を残すと説明しました。物体より灰のほうが普通軽いのでここまでは説明できます。しかし抜け出したフロギストンの実体についての解明はうまく出来ませんでした。その後近代化学の発展とともに元素の発見や分子の化学が進んだ結果,燃焼によって水や二酸化炭素など目に見えない気体に変化するとして理解され,物質変化を化学反応の方程式で表したり,さらに燃焼に伴う発熱を化学エネルギーから熱エネルギーへの変換と説明するようになりました。
燃焼は熱の発生を伴う激しい変化ですが,似たようなことを穏やかに行なうこともできます。食物を食べて分解し,その栄養素を吸収して身体の運動エネルギーに使うと最後は水と二酸化炭素になりますから,結果として燃焼をゆっくりやっていることになります。どちらも酸化反応ですが,ゆっくり分解したほうが途中でいろいろな中間産物が得られ,われわれは必要に応じてそれを組織細胞に送って複雑な生体機能を発揮させています。グルコースやアミノ酸など基本的なものから,ビタミン,ホルモンなど高度の機能性物質が体内で利用されています。
このように燃焼と分解は共通面のある化学現象ですが,分析化学では目的によってうまく使い分けています。燃焼は物質を酸素と急激に化合させて,水や二酸化炭素など最終酸化物まで持ってくることを普通言いますが,原子吸光分析ではもっと高温の炎を得るため酸素の代わりに一酸化二窒素(亜酸化窒素)で燃焼させることもあります。有機元素分析ではそれぞれの構成元素が決まった最終物質になる必要があり,このため800℃から1000℃あたりの酸化条件を与えています。同時に燃焼を促進する反応触媒やその中での滞留時間などが長い間検討され,結果が現在に生かされています。一方分解は物質を構成する分子内の化学結合を切断することで,より小さい分子になりますが,一般的には溶液内で試薬と反応させて新しい化学物質に導きます.水も試薬と考えれば加水分解もこれに含まれます。ケルダール分解やアセチル基,メトキシル基の定量などわれわれに身近な定量的分解反応もありますが,水質試験の化学的酸素要求量(COD)や脂肪のけん化価の測定など,一応定量値は出ますがこれでよいのか手応えのはっきりしない反応もあります。特殊なものとして熱分解ガスクロマトグラフィや質量分析があり,ここでは試料物質を熱や電子線で直接分解し,生成したフラグメント(断片)のスペクトルを解析しますが,それぞれで独自の技術部門を形成しています.本稿ではわれわれが日常接する有機物の燃焼と分解の化学について述べたいと思います。
2-1.燃焼の化学
燃焼は酸素や空気の中で行なわれる現象ですが,常温では普通ほとんど進行しません。燃焼には最初のきっかけが必要で,マッチやライターを用いて燃えやすいものの一部を加熱し,そこから燃え広がらせます。燃え広がるのは燃焼熱が伝播して加熱域を広げるからで,ここでは酸化の連鎖反応が進展します。有機物が燃えやすいのは炭素骨格に水素が普通結合しているからで,有機物を部分的にでも加熱すると炭素―水素間の結合が切れ,ラジカル場 (radical site) を作ります.ラジカルは結合を引き離された不安定な場所ですから,そこに酸素分子が近づくとこれと結合して過酸化物を形成します.有機物をRHの化学記号で表すと反応式は,
RH+△= R・+H・(△は熱エネルギー)
R・+O2 = R-OO・+△
H・+O2 = H-OO・+△
となりますが,出来たものは何れも不安定なラジカル物質ですから,遊離している水素ラジカルH・と結合してR-OOHやH-OOHとなったり,そのときの反応熱で炭素鎖が切れてもっと低分子の酸化物となったりします.アルデヒド,ケトン,有機酸から二酸化炭素,水などが生成します。酸素が過剰にあれば反応熱が蓄積して温度が上がり周囲に伝播して急速に燃焼域は広がります。
酸素分子が反応しやすいのは,2個の酸素原子の特殊な結合状態が原因します1)。酸素原子は [1s22s22p4] の電子配置を持っていますが,内側の1s,2s軌道の電子はよく安定しているものの,外側の2p 軌道の電子4個が少し不安定な状態になっています .この様子は分子軌道で図1に表されていますが,要するに右と左に描かれた原子軌道において4個の2p電子が2px, 2py, 2pzの3軌道に入るため,2pz軌道だけに2個スピン(自転)を逆平行にして磁場を打ち消すように入り,他の2軌道にはそれぞれ1個づつ孤立して入って います.このような左右の酸素原子が結合して中央の分子軌道を形成してもこの孤立した電子のスピンは解消せず,いわば酸素分子はラジカル場を2個所持った ビラジカル・O-O・の性質を持っています。このビラジカルは不安定で,相手の物質から水素を引き抜いたり,不飽和結合があれば直ちに結合してエポキシ基 を形成するなど,自身の不安定性を打ち消すように働きます。
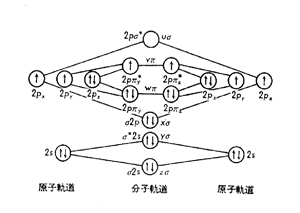
図1. 酸素の原子および分子軌道
ラジカル物質である酸素の高い反応性によってわれわれは有機物を短時間に完全燃焼させることができます。温度を高くし,酸素を十分供給すれば,究極の酸化物である二酸化炭素と水になります。この簡単といえる原理はしかし実際問題として過去にいろいろな曲折がありました。高い温度の得られなかった時代,酸化触媒を用いて完全酸化をはかる工夫がされましたが,その後自由に高温度が得られるようになって,逆に高過ぎて燃焼管と充填物の融着や生成ガスの熱解離など新たな問題が生じてきました。
3-1.燃焼触媒
有機物を燃焼させるといっても素直に燃えるものばかりではなく,難燃性のものや硫黄,ハロゲン,リンなどを含むものがあり,一部には金属元素を含むものがあって,元素分析の初期の段階からこれらの対策を強いられました。ガラス管に酸化銅とクロム酸鉛の混合物を入れて加熱し,酸素を通じながら有機試料を徐徐に気化して追いこむという方法は,最初誰が考えたのかよく分かりませんが,最初に分析法を形として残したリービッヒ (J. Liebig, 1803-1873,図2) は当然のようにこの方法を使っています。硫黄酸化物はクロム酸鉛で,ハロゲンは銀線を詰めて除去しました。

図2. J.Liebig
有機試料を気化するといっても当時は試料量が数グラムありましたから,キャリヤーガスの酸素だけで燃焼することは出来ず,酸化の主体は酸化銅との接触で行なわれました。銅は貴金属の性質を持っているため,酸化銅は容易に酸素を放出して単体まで還元しますから,酸化容量が甚だ大きい特長があります。酸化銅は早くから線状のものが用いられましたが,これは純粋な金属銅が電線用として工業用に作られていたからで,適当な長さの銅線を束ねて電気炉に入れ,酸素を通じながら750~800℃に保ち酸化します2-p.136)。酸化銅は金属銅より体積が大きくなりますので膨張して亀裂が生じ,その隙間から継続的に酸素が供給され芯まで酸化が進みます。この酸化銅を取りだし,冷後軽く砕いて2~5mmの長さに揃えたものが線状酸化銅として市販されています。
酸化銅はCuOで表されますが,実際は亜酸化銅Cu2Oが大部分で,残りが酸化銅と少量の金属銅です。仮に全部が亜酸化銅であったとして,1グラム当たり供給できる酸素量はO/Cu2O = 16/143.1 = 0.112 グラムです.酸素O2の体積にすれば0.112×(22.4/32) = 0.0784リットルとなります。リービッヒのマクロ分析の時代は多分150グラムほど充填していたと思われますが,微量分析になると約50グラムとなり,後者の場合酸素量は体積に換算して3.9リットル前後になります。もっとも亜酸化銅の酸素が全部燃焼に使われるはずはないので,これは単なる計算上の上限数値に過ぎません。
一方試料の燃焼に必要な酸素量はどのくらいかと言うと,例えば安息香酸を採取した場合,
C7H6O2 + X O2 = 7CO2 + 3H2O
ですから,安息香酸1モルあたり外部から取りこむ酸素のモル数は X = 7 + 1.5 – 1 = 7.5です. 分子内にO2がありますので1を差し引いてあります。安息香酸の分子量は122.12ですから,1 mg当たり必要な酸素量は7.5/122.12ミリモルと言うことになります.体積に直すと22.4×{7.5/122.12} = 1.37 mlですから,試料量が3 mgのとき4.12 mlになります。水素含量の多い炭化水素や,逆に窒素や酸素含量の多いものでは必要酸素量も違いますが,上の計算で亜酸化銅の酸素供給能力は余るほどあります。
CHN分析では試料を気化して酸化銅に通じることで燃焼に必要な酸素は十分と分かりますが,試料が炭化したりタール状になったりするとやはり酸素ガスを通じなければなりません。ここでの酸素は試料の完全気化が目的ですから,必要最小限に止めるのが得策です。ポンプシステムによるCHN分析計では3),10%の酸素を含むヘリウムを5分間供給していますが,ポンプの引きこみ速度が30 ml/minのとき,酸素の全通過量は15 mlということになります。試料の燃焼に必要な酸素量の3倍以上はありますが,流通系ですからそううまく気相中の酸素を利用できるはずはないので,多分試料のほとんどが酸化銅との接触で燃焼が進行します。酸化銅はこの時還元されますが,同時に含窒素試料から発生する窒素酸化物(NO, N2O, N2O)をその場でかなりの程度N2まで還元します。窒素酸化物は酸化銅など金属酸化物と一時的に結合して保留される性質があり,燃焼と同時に還元しておけば酸化銅内での窒素酸化物の保留も随分軽減されます。
試料の燃焼時還元された金属銅はあとでキャリヤーガス中の酸素で酸化されますが,いわば酸化銅は酸素を出したり入れたりする銀行のような役目を果たしています。このような作用が円滑に行なわれるためには,銅と酸素の結合力が手ごろである必要があります。一方高温に耐えるためには,銅と酸素の結合力がある程度大きくないといけませんが,かといって大き過ぎては酸素の放出がうまく行きません。金属酸化物の安定度のパラメータとして生成エネルギー(kJ・mol-1)がありますが,いくつかの例を挙げると4),
| CeO2 | Cr2O3 | Cu2O | Fe2O3 | MnO2 | MoO3 | WO3 |
| -1089 | -1140 | -169 | -824 | -520 | -745 | -843 |
となり,負の値が大きいほど沢山生成時エネルギーを放出してしっかり結合しているので,高耐熱性であり,その分酸素の放出が難しいということになります。酸化銅はこの中では最も結合力が小さく,容易に酸化,還元ができることを示しています。古くから重宝された理由がこれで分かります。ただし上に挙げた数値は金属酸化物の多分一番安定性の高いもので,当然もっと高次の酸化物もありますから,それらの段階と段階の間で酸素の放出,吸収もあってしかるべきです.酸化セリウムや酸化タングステンを酸化触媒に用いる例もあり,酸化物の段階間の遷移で酸素の供給を受けていることが考えられます。
酸化銅の融点は1200℃あたりですが,上に挙げた金属酸化物の中では一番低いので,あまり高温で長期間使用していると石英管と融着します。900℃が使用の上限と考えたほうが安全です。古い話ですがコレステロールの角メチル基が分解できないので,いろいろな酸化触媒を加えた研究がありましたが5),当時は燃焼管が粗悪で高温にすると曲がることから,せいぜい700℃か少し上程度の温度であったからと思います。現在のように850~900℃であればコレステロールはじめ普通の有機物は完全酸化できるはずです。
3-2.ヘテロ元素への対応
ハロゲンや硫黄分析を燃焼管法で実施しようとすると,金属酸化物を燃焼触媒に用いることができないので,キャリヤーガスを純酸素とし,白金を触媒としました。白金はいろいろな気体を吸着する性質があり,化学反応の触媒にもよく使われますが,使用形態も板状,網状,担体表面に分散したコロイド状と異なっています。有機元素分析では燃焼管の内径に合った,断面星形の細長い板状構造のものや網を丸めた円筒形のものを挿入しましたが2-p.139)(図3),
図3. 白金網によるハロゲン,硫黄試料の燃焼
高温で酸素を吸蔵する性質があり,酸化銅ほどではありませんが,多少酸素の貯留の役目を果たします。しかしこれでは酸素不足なので,できるだけキャリヤーガスの酸素と試料分解ガスとをうまく混合して燃焼管を通過させるのがコツで,分析技術者の腕の見せ所でした。
有機試料に不揮発成分があれば灰として残ります。新しい白金ボートを使いはじめると,間もなく汚れが目につくようになりますが,何か灰分が残っている証拠です。分析結果に響くほどではないので無視していますが,アルカリ分が蓄積すると炭素値に影響する可能性があり,時々硝酸で洗浄しなければなりません。ひどい汚れになった時は酸性硫酸カリウムを試験管に入れ,ガス炎で溶融して,その中に浸漬し,水洗すれば汚れは除去できます。灰分の存在が目で認められたときはボートを取り出し,冷却後はかりで計量して増量をチェックします。時間を置いた差の計量ですからあまり正確な数値と言えないのですが,5μg以上増量があれば残留灰分と考えてよいでしょう。灰分がボートの中に全部残ればよいのですが,恐らくボートの周辺に飛び散り,燃焼管内壁や充填物表面に付着します。ボートの増量を試料量から差し引いてCHNなど元素分析値を計算し直すのは理にかなっていません。灰分の化学形が分かっていないからです。このときはボートの増量を試料の不純性として試料提供者に回答すべきです。
ナトリウムやカルシウム塩などと分かっている試料は,これらのアルカリ分をタングステン酸塩として固定します2-p.14)。この処理をしないと燃焼後炭酸ナトリウムや炭酸カルシウムを形成して分解に時間がかかり,所定の燃焼プログラムについて行けないことが起こります。実際には元素分析用酸化タングステン約50 mgを目分量でボートの中の試料に振りかけます。燃焼によって生じるタングステン酸塩は炭酸を追い出し,高融点でボートの中に残ります。厄介なのはリンを含んだ試料で,特にタンパクや生体物質に多いものですが,ポリリン酸の形で高温部を通り抜け,低温部に付着します.燃焼管を出た部分や場合によっては検出器の方まで来る可能性があります。水を保留しますから燃焼管と還元管,さらに検出器までの連結管は時々外して水で洗浄すべきです。
分析能率が向上し,一日に多数の試料を燃焼するようになりましたが,試料中の不純物の累積も問題になります。燃焼管の内壁の汚れである程度状態は掴めますが,不純物が充填物の表面を覆うと燃焼効率は下がり,それがアルカリ金属などであれば二酸化炭素の通過を妨げます。現在は分析装置が電子化されて標準試料で感度を決めていますが,こういった機能低下を読み込んだ感度を使うことになりますので,当面はよいとしても長続きはしない可能性があります。試料提供者に物質精製を徹底してもらうことと,燃焼管充填物の更新を早い目にすることが大切です。古い時代には一回の分析に一時間ほどかけたので,失敗は痛手でした。試料を作る方でも再結晶を繰り返し,融点が変わらぬことを確認してから提出していました。近年は再結晶法より液体クロマトのフラクション(分画)を取り分け,溶媒を飛ばせた残留分を提出する人が増えてきました。一見して飴状のものですが,とりあえず分析に出して理論値に合えばよいという安易な考えのようです。僅かな不純物を含んでいますが,主成分は目標物でしょうから理論値から少し外れた分析値が得られます。分析依頼者からは核磁気共鳴や質量分析のスペクトルデータが目標物に一致するとクレームが来ますが,これらのスペクトルデータは僅かな不純物を検出しないので,議論にはなりません。
以上は燃焼管法による有機試料の燃焼について述べましたが,1950年代突然現れたシェニガー(W. Schoeniger)の酸素フラスコ燃焼法はハロゲン,硫黄分析の燃焼形態を一変させました6)。その手軽さと信頼できる分析結果で,それまでの燃焼管法はたちまち姿を消してしまいました。酸素を充たした三角フラスコの中で,試料を包んだ濾紙を燃焼させるというアイディアはまことに卓抜しています。酸素雰囲気中で濾紙は1500℃の炎となり,この中で数ミリグラムの試料は完全燃焼します。閉鎖されたフラスコの中で分解ガスは,あらかじめ入れておいた吸収液に定量的にとりこまれ,あとは滴定や吸光光度法で測定できます。JISや薬局方にも収載され,有機元素分析の枠を越えて品質管理や公定試験に広く応用されています。酸素フラスコ法の詳細については別項「フラスコ燃焼法と密閉反応器」に記述されていますので参照してください。
4-1.元素の定量
燃焼も分解の一種ですが,高温の雰囲気で行われる気相反応ですから,電気炉や生成物の誘導,捕集にいろいろ工夫が必要です。液相で試料を化学的に分解すると反応生成物は液相に溶け込んでいることが普通で,定量分析には成分の損失が無くて有利な面があります。ただし試料に比べて圧倒的に多い反応試薬が残っていますので,目的成分の測定プロセスに障害がないか検討が必要です。一番確実な方法は反応後沈殿試薬を加えてろ過し,沈殿の質量を計量したり,目標物質が揮発性であれば蒸留したりすることですが,いずれも面倒な手作業なのであまり利用する人がなくなりました。しかし反応液を用いて目的成分の電位差滴定や吸光光度測定をすることは比較的手軽で今でもよく使われています。
湿式分解法の古いものではカリウス(L. Carius)法7)とケルダール(J. Kjeldahl,)法8)がありますが,前者はガラス封管の中で試料を発煙硝酸と共に加熱し,分解液からハロゲンイオンや硫酸イオンを測定しました(図4)。加熱の間高圧になるので肉厚のガラスを用いましたが,封管のさましが悪いとひずみが残り,爆発することもありました。一旦爆発すると発煙硝酸が飛び散り,後始末が大変で,現在では全く使われなくなりました。これに比べると後者は開放された試験管に試料と濃硫酸をいれて加熱するので危険はなく,現在でもアミノ態窒素の定量によく使われています。ただし反応中硫酸ミストが出るので,精密な分析機器に障害が起こらぬよう,ドラフトなど腐食性ガスの排出設備が必要です。難点はアミノ態窒素以外,すなわちニトロ基やアゾ基などには定量性がないので,試料を還元してアミノ態窒素に変換する前処理が必要になります。しかし面倒な化学操作ですから,そうまでしてケルダール法にこだわる人はあまりないでしょう。
図4. 封管の作り方
日本薬局方では一般試験法の中に「窒素定量法」の項目があって,装置や操作法が規定されています。図5が装置ですが,分解フラスコAの中であらかじめ試料と濃硫酸,硫酸銅,硫酸カリウムを加熱して試料中の窒素を硫酸アンモニウムに変換します。硫酸銅は変換触媒で,硫酸カリウムは混液の沸点を上昇するためのものです。液が炭化すると黒ずんでくるので,過酸化水素の数滴を加え液を澄明にします。分解フラスコを装置に取り付け,Fから水酸化ナトリウムを加えるとアンモニアが発生しますから,丸フラスコBから水蒸気を送って受器Kに凝縮液を滴下させます。Kにはホウ酸液を入れておきアンモニアをホウ酸アンモニウムとして捕集します。薬局方ではブロモクレゾールグリーン:メチルレッド混合液を指示薬として0.005mol/L硫酸標準液で滴定(緑_赤紫)しています.
(0.005 mol/L) H2SO4 1 ml = 0.14007mg N
ホウ酸は弱酸ですからこの滴定の障害にはなりません。薬局方では目視法が規定されていますが,一般分析ではpH電極による電位差滴定法やインドフェノール吸光分析法9)を採用したほうが便利でしょう。
ケルダール分解法では濃硫酸で分解し,有機窒素をアンモニアとして水蒸気蒸留で分離していますが,ヒ素,カドミウム,鉄,リンなどの定量にも同様の湿式分解法を用いることができます。多くは原子吸光法で各元素を測定しますので,分解液に硫酸を用いるとネブライザーの噴霧効率が悪くなり,このため硝酸や過酸化水素が使われるようになりました。ただし沸点が硫酸のように高くないので,テフロンの反応容器をこれに緊密なステンレスの外装に密閉し,加熱装置に入れて分解することが提案されました10)。ユニシールの名称で市販されましたが,内容積70 mlで分解液は1/3ほど入れ,140 ℃の加熱装置で4時間反応させています。テフロンは化学的に不活性なのでフッ化水素によるケイ酸塩の分解にも利用できます.ただしテフロン壁はあまり厚くないので使用とともに変形し,蓋の密着性が悪くなりますので時々新しいものに交換が必要です。
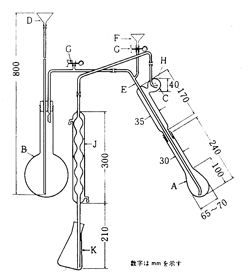
図5. 薬局方窒素定量装置
最近は金属外装なしで,図6のように硬質のテフロン容器に分解液を入れ,さらに肉厚のプラスチック容器に収容し,マイクロウエーブで外部から加熱する方式が広がっています11)。初期の段階では家庭用電子レンジを使っていましたが,現在は専用の分解装置がいろいろ市販されています.マイクロウエーブ(2450 MHz)はプラスチック容器を透過しますが,水性の分解液にはよく吸収されますので放射エネルギーが熱に変わります.もともと極性を持った水分子の回転周波数に同期させるもので,本当は2000MHzがよいのですが,工業周波数の制限があり,上記の周波数が実用されています。容器は上部のスクリューねじで締めますが,反応中は高圧になりますので器具を使ってしっかり閉じなければなりません。12個の反応容器が放射状に取りつけられ,回転しながらマイロウエーブ600 Wを投射します。反応温度を制御するためにマイクロウエーブを間歇的に加え実効電力300 Wほどにする工夫がされています.分解液は200~300℃くらいになり,数分から20分で分解は終了します。温度が高いことが反応速度の速い理由ですが,もう一つマイクロウエーブの特性も寄与しているようです。水や酸など極性物質のマイクロウエーブによる分子回転がありますから,単なる熱エネルギーによる分子衝突とは反応効果が違うのだと言われています。微量分析にはまだあまり実用されていないようですが,環境試料や生物試料を原子吸光など機器分析にかけるための前処理には繁用されるようになりました。
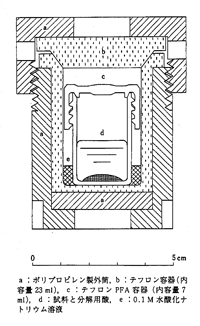
図6. マイクロウェーブ加熱容器
分解液を使わないので湿式とは少し言い難いのですが,試料と過酸化ナトリウムをニッケル製ルツボに入れ,ガス小炎で溶融加熱するパールボンブ(Parr-Bomb)法があります12)(図7)。試料ごとに金属のキャップをスパナで締め,密閉して加熱分解し,あとで開放して溶融物を洗い出すので面倒な点はありますが,分解は確実です.有機ケイ素など分解し難い試料の分析に使われましたが,その他ハロゲン,硫黄などヘテロ元素の定量に汎用できますので,本当は推奨したい方法ですが,何でも機器化の現代相にはあまり人気が出ないようです。
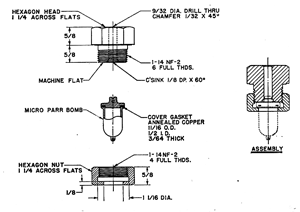
図7. Parr-Bomb分解容器
4-2.原子団の定量
赤外スペクトル法が出来てからメトキシル基や水酸基など原子団の定性は容易になり,さらに核磁気共鳴の機械が普及して化学構造の中での存在場所や数まで読み取れるようになりました。かつてはどこの有機化学実験室にもあった原子団定量のガラス器具はほとんど見られなくなり,やり方も知らない人が増えています。電子情報から未知物質の推定構造式を組み立て,試料の元素分析値が推定構造の理論値に一致すれば終わりとするケースが多いようですが,本当は原子団の定量値も確かめてほしいものです。原子団定量が公的に規制されているのは日本薬局方で,メトキシル基定量法と油脂試験法に収載されています.前者は軟下剤やローション液の粘りを増すのに使われるメチルセルロースが対象で,セルロースの高分子鎖に存在する水酸基のうち,どのくらいの割合でメトキシル基が置換しているかを管理します.後者はカカオ脂,オリブ油,ラノリンなど動植物の油の品質を,酸価,エステル価,水酸基価,ヨウ素価などの項目で試験します。
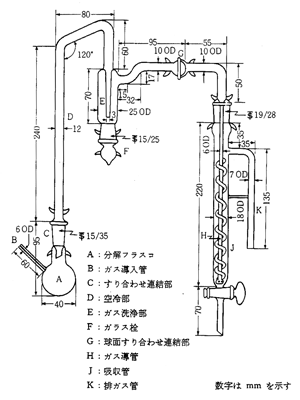
図8. メトキシル基定量装置
メトキシル基の定量はツァイゼル(S. Zeisel)法が基本で13),試料とヨウ化水素を図8の分解フラスコに入れ,小バーナーで加熱します。窒素または二酸化炭素を通じて生成するヨウ化メチルを気相に追い出し,この後赤リン懸濁液で洗浄し吸収管に送ります。吸収管には酢酸―酢酸カリウム混液を入れ,少量の臭素を加えておきます.ヨウ化メチルは酸化され,ヨウ素酸となります。
ROCH3 + HI = CH3I + ROH
CH3I + 3Br2 + 3H2O = CH3Br + 5HBr + HIO3
連結管のボールジョイントを外し,水を加えて数回吸収液を共栓三角フラスコに流し出します。過剰の臭素で赤みが残っていますので,ギ酸を無色になるまで加えます。この後ヨウ化カリウムと希硫酸を加え,遊離したヨウ素を0.05 mol/Lチオ硫酸ナトリウム液でて滴定します。
HIO3 + 5HI = 3I2 + 3H2O
3I2 + 6Na2S2O3 = 6NaI + 3Na2S4O6
0.05 mol/L チオ硫酸ナトリウム1ml = 0.5172 mg CH3O
この滴定はフィーベック-ブレッヘル(F.Viehbock-C.Brecher)法と呼ばれ14),ツァイゼル原法で用いられた沈殿重量法より簡単で正確なため推奨されています。それはヨウ素酸1モルから6原子のヨウ素ができるからで,いわば6倍に化学増幅がされています。注意すべきは臭素を使うことで,臭素の瓶は滴定現場の近くに置かない事です。瓶から揮発した重い臭素の気体が実験台の上を漂い,滴定終点で余分なヨウ素を発生させて誤差を生じます。また最初の試料の分解で,物質がヨウ化水素に溶けにくいときはヨウ化水素,フェノール,プロピオン酸で構成されたキルステン(W. Kirsten)の混液が推奨されます15), 2-p.142)。
アスピリンやアセトアニリドはサリチル酸の水酸基やアニリンのアミノ基にアセチル基が脱水置換したものですが,酸によって加水分解すれば酢酸を生じ蒸留することができます。最初に不揮発性の強酸による分解が必要ですが,これにはp-トルエンスルホン酸が適当です。酢酸など有機酸の基体をアシル基RCO-といいますが,蒸留できる程度の大きさの酸でないとアシル基の定量はできません。
RCO-OR’+ H2O = RCOOH + R’OH
RCO-NHR” + H2O = RCOOH + R”NH2
アミン類に亜硝酸を作用させるとアミノ基が酸化されて定量的に窒素となりますが,この窒素ガスを体積または圧力として測定する方法が1911年オランダのバンスライク(D.D.Van Slyke)によって考案され,以来生化学分野で大きな発展をとげました16)。アミノ酸やタンパク質研究には必需品で,バンスライク法はそれ自身独立した分析分野になった時代がありました。現在は質量分析や核磁気装置が代わりの役目を果たすので研究室からは姿を消しつつありますが,病院検査室や環境分析室などにはまだ現役で使われています。亜硝酸は水中で不安定な物質ですから,実際には使う前に亜硝酸ナトリウム溶液に酢酸を加えて作ります。脂肪族一級アミンと反応させると常温で窒素を生じます。
NaNO2 + CH3COOH = HNO2 + CH3COONa
RNH2 + HNO2 = ROH + N2 + H2O
バンスライク法に関連してアゾトメトリーという方法が金沢大学の岩崎 憲教授によって考案され,生化学領域で超微量分析に応用されました17)(図9)。アミノ基を持つ試料の多くはアルカリ性で次亜臭素酸によって酸化分解し,窒素ガスを生じますから,ガス体積を毛細管のようなビュレットに集め測定します。
2RNH2 + 3NaOBr = 3NaBr + 2ROH + H2O + N2
ビュレットの中を二酸化炭素で置換し,試料液とアルカリ試薬を導入すると真空になりますから,常圧に戻し目盛りで体積を読みますが,少々手際よく操作しなければなりません。詳しくは文献を参照してください2-p.270)。アミノ基以外の窒素に対しては多少工夫が必要ですが,最終的には窒素ガスにして定量できます。
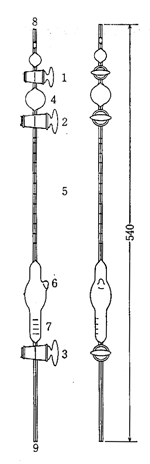
図9. 超微量アゾトメータ
5-1.おわりに
分析操作の最初にやるべき燃焼または分解について解説を試みました。酸化は破壊的な化学操作で,われわれの実験室では元素または簡単な無機物に到達します。窒素,二酸化炭素,水など統一した定量形が得られるので処理も簡単です。もっと高温の燃焼法も原子吸光やプラズマ発光に使われていますが,ここではイオン種が生成して信号が基底種のものかイオン種のそれか議論が起こります。1000℃以下の電気炉で元素分析をやっているわれわれはこの点迷いがなくて幸いです。しかし二酸化炭素は800℃以上になると僅かですが解離を始め,もし還元銅のような酸素を除去する金属があると著しく一酸化炭素を発生させます。還元銅を通過した二酸化炭素はもう一度酸化銅などで酸化し直す必要があります。
分解は化学結合を切って低分子化させますが,必要にして十分な化学エネルギーを加えなければなりません。有機物を過マンガン酸カリウムで処理するとあちこちで切断がおこりますが,決まった切れ方をしないので定量分析に使えません。アルデヒドやカルボン酸などいろいろな形で分解するので,既述のように水質汚染の指標に過マンガン酸カリウム液の消費量で化学的酸素要求量(COD)を測定していますが,お役所的な取り決めで分析化学的にはもう一つすっきりしません。これに比べるとメトキシル基やアセチル基の定量は正確に目標を捕らえています。機器分析の発達していなかったころの有機化学者は日常このような原子団の定量を繰り返し,化学構造の実態を体感していました。赤外,核磁気,質量分析の普及で有機化学者はコンピュータから出てくるデータの解析で物質の化学構造を推定してしまうので,これら原子団の定量法を知らない人が増えています。電子情報をマニュアル通り解析して物質の全体像を組み立てても,果たして本質に迫っているのかどうか,群盲象を撫でることにならなければ幸いです。
6-1.参考文献
- 森野米三,藍原有敬: 現代物理化学講座2 “原子の構造と化学結合” p.114, 東京化学同人 (1966).
- 日本分析化学会有機微量分析研究懇談会編: “有機微量定量分析”,p.136, p.139, p.141, 南江堂 (1969).
- K. Hozumi: Microchem. J.,10, 46 (1966).
- 日本化学会編: “化学便覧,基礎編 II”,p.305, 丸善 (1984).
- 元素分析懇談会編: “最近の元素分析法”, p.134, 南江堂 (1955).
- W. Schoeniger: Mikrochim. Acta, 1955, 123.
- L. Carius: Ann., 116, 1 (1860).
- J. Kjeldahl: Z. Anal. Chem. 22, 366 (1883).
- L. T. Mann: Anal. Chem. 35, 2179 (1963).
- 内田哲男: ぶんせき,1986, 9.
- 小島 功: ぶんせき,1992, 48.
- S. W. Parr: J. Am. Chem. Soc., 30, 764 (1908).
- S. Zeisel: Monatsh. 6, 989 (1885).
- F. Viehbock, C. Brecher: Ber., 63, 3207 (1930).
- W. Kirsten, S. Ehrlich-Rogozinsky: Mikrochim. Acta, 1955, 786.
- D. D. Van Slyke: J. Biol. Chem., 9, 185 (1937).
- 岩崎 憲,大月 理,中島誠一: 十全会雑誌,42, 2125 (1937).