



TOPICS
CHNフォーラム
第一部 センサーの知識
- 1-1.はじめに
- 2-1.光のスペクトル
- 2-2.紫外可視光線のセンサー
- 2-3.光測定の基本
- 2-4.赤外線のセンサー
- 3-1.ガスセンサーの種類
- 3-2.熱伝導度検出器
- 3-3.ブリッジ出力の漂動
- 4-1.ボルタンメトリー
- 4-2.電位差測定
- 4-3.電気伝導度測定
- 5-1.おわりに
- 6-1.参考文献
- 関連製品
1-1.はじめに
人間には五感があるとされ、目、耳、鼻、舌、触覚で周囲の状況を感じ取って日常の生活や危険の判断に役立てています。特に目と耳は離れたところで何が起こっているかを知るのにすばらしい情報源です。光は一秒間に30万Km(地球を7周り半)もの速度で情報を伝達してくれますが、これを利用して古くは地平線の彼方に上がったのろしの煙で外敵の来襲を知ることができました。野球ではピッチャーの投げた時速150 Kmを超える高速球を目で捕らえ,細いバットで正確に打ち返しますが、光の速度がボールよりはるかに速いから出来ることです。音の速度は光に比べるとはるかに遅く、せいぜい秒速340mほどですが、それでもわれわれの行動空間では十分高速で、オリンピックスタディアムでは何百人ものマスゲームが音楽に合わせて一斉に演技できます。
人間に限らず生物は環境に適応して生命を維持するため、周辺情報をキャッチする感覚器官を備えています。基本的には物理的な刺激か化学的な反応を微弱な電気信号に変え、このあと信号の解析により遺伝子的な自発命令(自律神経系)か後天的な判断(大脳皮質系)により行動を起こします。後天的な判断は知識と経験の深さによって質が決まりますが、人間は長い期間教育を受けますので、他の動物に比べればはるかに賢明な判断を下すことができます。信長の桶狭間の勝利などは情報と決断の見事な融合によるものでしょう。
情報をキャッチして電気信号に変換する道具をセンサーと呼んでいますが、電子回路に組み込まれたものだけでなく、結構生物は神経回路に組み込んで古くから同じ目的を達してきました。むしろ生物が進化の過程で獲得した機能を人間が道具で真似したのだというべきでしよう。化学の研究を始めたのは人間ですから、人間に備わった感覚器をまず利用したのは当然で、手近な目、鼻、舌を使って物質の本体を探り出そうとしました。そのうち時代の進歩とともにこれらが理学的な道具に代わって現代の姿になっています。
センサーの役目としては、目的物が在るか無いかを判定する場合もありますが、在ることが予測されていて、その量が重要である場合もあります。火災報知器などは前者に属しますが、雨量計などは後者に属します。分析化学の分野では定性分析と定量分析があり、目的の面では別になっていますが、使うセンサーは共通ということもあり、この場合は道具の規格や使い方で定性分析向きや定量分析適格ということになります。
近年は分析機器がコンピュータ制御になり、分析化学反応が行われている本体よりも電子装置を搭載した周辺機器の華やかさのほうが目を惹きますが、根本はセンサーから出た微弱な電気信号を拡大して、画面やプリンターに表示させているので、センサーの出力が正しいことを前提としてわれわれに情報を届けています。従って本当にセンサーの出力は正しいのか、また初め正しかった出力が時間の経過とともに今は間違った方向にずれてはいないのか、絶えず関心を持っていなければなりません。故障の修理などは専門家に委託するとしても、日常の僅かな気配りでセンサーを快適に作動させることに繋がることもあり、分析情報の源泉とも言うべきセンサーについてある程度の知識と扱い方は心得ておくべきです。本稿ではセンサーについて常識的な事項と忘れがちな問題を解説したいと思います。
2-1.光のスペクトル
虹の七色は太陽光線が空中の水滴で屈折して反対の方向に出て行くとき、色によって角度を変えることから起こる現象ですが、われわれの目は赤から紫までの波長しか感じないので、この世は七色で彩られていると思っています。ピカソやゴッホのような天才でもこの範囲の色しか使っていません。光の本体は電磁波で、電子の激しい振動があるとそのエネルギーが空間の歪となって波状に広がり進んで行く現象です(図1)。一秒間の振動数νが大きいほど波の保有するエネルギーも大きくなりますが、波の進行速度は一定なので、隣どうしの波の間隔すなわち波長λが短くなります。従って波長が短いほど光のエネルギーは大きく、波長が長いほどエネルギーは小さくなります(図2)。いろいろな波長が混ざり合った光の場合、横軸に波長を、縦軸に波長ごとの光の量を並べた図形をスペクトルと呼びますが、波長ごとの光の量は違うのが普通で、このとき図形は山や谷を持った複雑な曲線を描きます(図3)。なお物理学では電磁波は電子の振動だけでなく、一般的に荷電粒子の高速振動から発する放射エネルギーと定義していますが、われわれが普通扱うのは電子の振動によるものです。
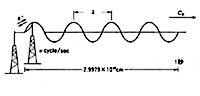
図1 電磁波の進行
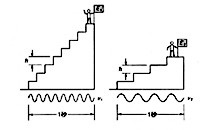
図2 光のエネルギー (h:ブランク定数)

図3 過マンガン酸カリウム液の吸収スペクトル
具体的には原子や分子が相互に激しく衝突したり、他から高エネルギーの電子や電磁波hνが飛び込んできたりして、それらの軌道電子が安定状態(基底状態)から高いエネルギーをもつ軌道に押し上げられ(励起状態)、次の瞬間元の安定軌道に落ちてくるとき、軌道間のエネルギー差を光として空間に放出するものをいいます(図4)。このエネルギー差は光として放出する以外、熱の形で失われるものもありますが,電子軌道論ではこれらを総括して軌道間のエネルギー遷移と呼んでいます。太陽表面は約6000度の水素ガスに覆われていますが、この温度で水素分子は相互に激しく衝突し、そのときに発する光を太陽の恵みとしてわれわれは受け取ってきました。
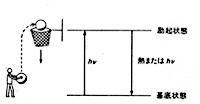
図4 物質のエネルギー遷移
人間の目に見える光の範囲は赤から紫までのいわゆる可視光線ですが、多少個人差があるようで、特定の色が欠けて見える人もあります。昆虫は人間の可視光線より波長の短い紫外線がよく見えるらしく、紫外線ランプで呼び寄せて電撃を加える誘蛾灯を作っています。爬虫類は逆に波長の長い赤外線を感じるセンサーを持っていて、動物の体温から発する赤外線をめがけて襲いかかります。可視光線というのは人間が自分の感覚で勝手に決めた波長範囲で、昆虫や爬虫類など他の生物は可視光線に違う波長範囲を主張するに違いありません。可視光線を中心として電磁波の広がりを示すと図5のようになりますが、全体から見た可視光線の範囲はほんの狭い領域であることが分かります。

図5 電磁波の名称と物質に対する作用
分析化学が主として目の感覚で行われていた時代は、可視光線の範囲の現象しか取り上げられませんでしたが、少し波長の短い紫外線や長い赤外線で測定すれば今まで見えなかった現象を知ることができます。砂糖と塩はどちらも白い粉末で、水に溶かしても無色で両者の区別がつきませんが、糖類は紫外線をよく吸収するので、昆虫などは独特の色として蜜を集めているのでしょう。このため人間は紫外線を感じるセンサーを作って観察することを覚えました。
光電管というものが20世紀の始めに発明されて、光を電流として取り出すことが出来るようになりましたが、これで人間はそれまで気が付かなかった自然現象を理解するようになりました。例えば日光浴で日焼けするのは人間に見える光のせいではなく、もっと短波長の紫外線が原因です。有機化合物は色の着いたものもありますが、圧倒的に無色または白色のものが多くあります。しかしほとんどのものが紫外線を吸収します。光電管を使えば無色の物質も着色物質と同じように認識できるはずです。どのような波長をどのくらい吸収するかは化合物固有の特質ですから、これを測定すると物質の特定や存在量を知ることができます。
可視光線より波長の長い赤外線も重要な情報源です。ストーブや焚き火の周囲にいると暖かいのは主として赤外線のせいですが,もう少し詳しくいうと赤外線を人間の皮膚が吸収して原子や分子間の振動を強め、これが熱エネルギーになるからです。ここでも生体を構成する有機化合物が赤外線を吸収するという原理が働いています。有機化合物はほとんど例外なく赤外線を吸収しますので、その吸収波長と吸収強度を調べると随分詳しい有機化合物の構造が推定できます。赤外線の検知には普通の光電管ではほとんど感度がないので,熱電対や半導体センサーを用いますが、測定装置はいまや有機化学者の分子構造推定のために日常道具として使われています。固体や液体物質だけでなく、気体の赤外線吸収も近年よく利用されます。都市部の二酸化炭素や自動車排ガスの汚染度は赤外吸収計が街頭で終日データを表示しています。
人間の能力を超えた自然界の観察が光センサーの力で可能となり、随分物質の化学情報が豊富になりました。分析化学者にとっては強力な味方で、大切に扱わなければなりませんが、それだけにどういう機構で作動し、どこまで使えるのかを知っておく必要もあると思います。
2-2.紫外可視光線のセンサー
前述の光電管はかなり古い歴史を持っていますが、原理的には図6のように真空ガラス管の石英窓から光を入射させ、セシウム-アンチモン合金を塗った金属板に当てます(1-p97)。セシウムはアルカリ金属で,原子軌道の一番外側に孤立した一個の電子を持っていますが、電子軌道の安定にはあまり寄与していないので、光のような外部エネルギーが加わると簡単に原子から飛び出します。金属板を陰極とし、その前に陽極を配置すると,飛び出した電子は陽極に流れ込み外部の電気回路に電流が流れます。負荷抵抗から光の量に応じた出力を得ることができます。
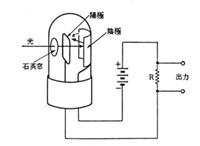
図6 光電管による光の検出
この出力信号は微弱なので増幅器で拡大する必要がありましたが、その後光電子増倍管が考案され、安価に量産されるようになって、直接大きな出力信号を得ることが出来るようになりました(1- p97)。図7に光電子増倍管の断面を示しますが、入射光ははじめ陰極に当たりいくらかの電子を放出します。この電子は数十ボルト正電位の第一ダイノードに引きつけられて加速を受け、ダイノード面に当たって放出電子(二次電子)の数を増やします。増えた二次電子は次のダイノードに引きつけられ、さらに高次の電子となって急速に数を増やします。10回ほどこのような増幅が行われると、最初の光電子は百万倍にも増え、増幅器なしで直接電流計の指針を振らせたり、デジタルカウンターの入力に取りこむことができます。光電子増倍管は感度がよいばかりでなく、光量と出力の比例関係(直線性)も良好で、定量分析には最良のセンサーです。
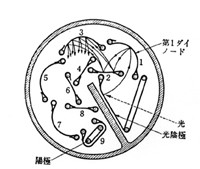
図7 光電子増倍管
光電子増倍管に比べると感度も劣るし、測定波長範囲も狭くなりますが、手軽な光センサーとして円板状チップを用いることもできます。光電導セルは石英板に蒸着した半導体の硫化鉛が光を受けると電気抵抗が下がることを利用していますので、簡単な直流電源を外部に取りつけると負荷抵抗から出力を取り出すことができます。太陽電池にも使われるシリコンチップは電源なしで電極から出力が得られますが、いずれにしてもこれら半導体のセンサーは光電子増倍管のように多目的には利用できません。
2-3.赤外線のセンサー 光測定の基本
定量分析は目的成分の存在量を知ることですが、存在量と測定する光の量との関係は単純ではありません。存在物から光が出てそれを測定するというのであれば,光の量はほぼ存在物に比例しますので、比較的簡単です。蛍光スペクトルやICP発光スペクトルなどはその例ですが、定量精度となるとある限度以上は期待できません。本来発光現象は基底状態の物質を励起状態に持ち上げて、それが再び基底状態に戻るときに放出するエネルギーのうち、光放射の分だけを見ているので、熱など無放射の分は何処へ行ったのか分かりません。このため発光量の再現性が悪くなっています。
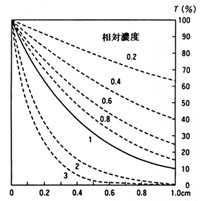
図8 セル内の吸光曲線
一方基底状態の物質に光を通すと決まった確率で光が吸収されます。しかし測定するのは透過した光の量で、この量は物質量に対応していません。このためベールの法則に基づいて透過光量と入射光量の比率Tを求めます(Tは%で示すことが多いのですが、計算の時は0.306など0~1の間の数値になります)。Tの対数は負の数になりますので、正の数値に反転するため-logTとし、これを吸光度Aとすると、Aの値が吸光物質の量に比例します(図8)。実際には目的物質を透明な溶媒に溶かし、セルに入れ光を通します。透過光量をIt、入射光量をIoとすると、セルの光路長lcmのとき試料濃度cとの関係は、モル吸光係数をεとして、
-log ( It /I0 ) = A = εcI
で表されます(2-p57)。あらかじめ濃度既知の溶液で濃度対吸光度の検量線を作っておけば、未知試料の濃度が求められます。現在の分光光度計は透過率のみでなく、演算機構によって吸光度も出力として得られますので、定量分析には便利になっています。
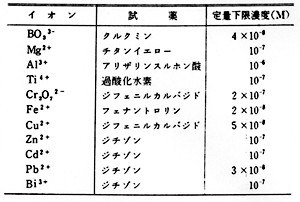
検量線の直線範囲は上限と下限がありますが、微量分析では上限を問題とするケースはあまりありません。下限は空試験値や発色物質の解離などがあって僅かに曲がることが多く、原点を通る直線にはなりませんが、標準液を用いて補正すればかなり正しい値が求められます。それでもこれ以下は無理というの定量下限濃度を表1の例に示しました。いずれにしても検量線の直線域では精度の高い定量分析が可能です.現在は機器の演算機能が発達して、検量線を記憶させておけば、直接未知試料の濃度を表示させることもできます。
表1 イオンの呈色反応と定量下限濃度
光電子増倍管は可視光線から紫外線領域にわたって優れた感度を示しますが、試料液をいれるセルの材質や、試料液の調製に用いた溶媒によっては測定波長範囲が制限を受けます。パイレックス製のセルは作りやすく、安価に入手できますが、紫外部で強い吸収を示すので可視部でしか利用できません。石英製のセルは少し経費がかかりますが、可視部から紫外部の200nmあたりまで透明ですから、専門の分析室では石英製を揃えるべきです。この様子は図9に示されています(2-p67)。
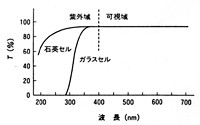
図9 セルの光透過性
もうひとつの波長制限は溶媒の光吸収にあります。有機試料は有機溶媒に溶けやすいので、試料液の作成には有機溶媒を用いますが、可視部では問題ないとして、紫外部では溶媒の光吸収が起こります。特に二重結合を持つ溶媒に顕著です。ベンゼン、アセトンなどはなるべく使わないほうがよいでしょう。エタノールやシクロヘキサンがよく使われますが、紫外線の吸収端が215nmあたりで、可視部からここまでの短波長域で測定が出来ます。これより短い波長では溶媒が紫外線を吸収してしまい、測定ができません。水の吸収端は200nmですから、無機物の試料ならば水溶液で測定するのが得策です。溶媒の吸収端を表2に示しました(1-p103)。
表2 溶媒の吸収端

高速液体クロマトグラフの検出器にも多くの場合紫外吸収計が使われていますが、溶離液の選択には成分の溶解性と溶離液の吸収端を考慮しなければなりません。成分の検出には波長を選ぶこともできますが、いろいろな成分に対応するため254nm付近に固定することが多いようです。もちろんこの波長で溶離液の光吸収がないことが条件です。また石油成分のようにこの波長で光吸収がないものでは示差屈折計を用いることになりますが、感度はかなり悪くなります。
2-4.赤外線のセンサー
赤外線のエネルギーでは光電管の陰極板から電子を放出させることができないので、薄い板状の熱電対に光を吸収させ、温度上昇を起電力として取りだしています。試料は粉末状にして臭化カリウムと混ぜ、打錠器で円盤状に固めたものを用います。赤外線を豊富に出すグローバーランプ(炭化ケイ素棒に電流を通じたもの)からの光を2本に分け、試料セルと参照セルに交互に導き、透過光を分光器にかけ、単色光を熱電対のセンサーに集めます。紫外可視スペクトルのように一定波長で定量することは普通ありませんので、4000 cm-1(λ=2.5μm) から600 cm-1(λ=16μm) まで自動的に走査してスペクトルを得ます。チョッパーで試料セルと参照セルの光を切り替えているので、熱電対の応答速度を速くする必要があり、このため極めて薄い白金板が受光器に使われます。
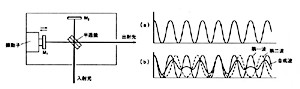
図10 マイケルソン干渉器と二波長の合成干渉波
最近は回析格子の分光器を用いないフーリエ変換赤外分光光度計(FTIR)が普及してきました(2- p81)。理論的には少し複雑な数理要素を含んでいますが、使うのはむしろ簡単になっています。図10のようなマイケルソン干渉計が主役で、半透鏡で2分された光がM1、 M2で打ち返されて出射光となります。M1は光軸方向に振動しているので、光路長が伸び縮みしており、M2からの反射光と合すると、光路差が波長の整数倍になるごとに図10-aのように強め合い山形を繰り返します。谷は1/2波長のずれで消光を意味します。波長が違うと点線のようになりますが、これらの合成波は図10-bのようになり、あとでフーリエ変換という演算をコンピュータで行えば、第一波と第二波のそれぞれの波長と強度を表示することができます。
フーリエ変換の赤外センサーには、熱電対の応答が振動鏡の速度について行けないので、もっと応答の速い重水素化トリグリシンスルフェート(DTGS)が用いられます。この薄膜が赤外線を吸収すると電気伝導度が変化することを利用したものです。フーリエ変換赤外分光法は、測定波長の全域が1回の振動の中に含まれるため、高速のスペクトル展開が可能で、また単色光を得るためのスリットがないので十分な光のエネルギーが検出器に加えられます。感度の高い検出がこれで行えるようになりました。
3-1.ガスセンサーの種類
われわれの生活は大気の中で行われていますが、酸素がないと文字通り窒息してしまいます。幸いどこにでもあるので特に酸素の検知能力は人間に備わっていませんが、炭坑やトンネル工事など特殊な場所では酸素濃度のモニタリングが必要です。ビルや住宅の煙感知器は現在どこにでも見られます。食品から発するにおいは食欲を促進したり、また腐敗の警報になったりしますが、このにおいはかなり高度の判断によるものです。野生動物になると獲物を追跡したり危険な相手を避けるのに、驚くほど敏感な嗅覚を働かせます。昆虫のフェロモンに到っては僅か1分子を辿って求愛行動に突進するといわれますが、人間にはとても真似のできないことです。
分析化学でもガス成分の検出はいろいろな局面で利用されています。最も重要なものはガスクロマトグラフに備えられた熱伝導度検出器です。無機、有機を問わずヘリウムより熱伝導度の小さい成分を検出し、感度の再現性もよいことから、定量分析に向いています。有機元素分析でC、H、 Nの同時定量を可能としたのもこの特性によるものです。
その後有機成分に特別に高感度の水素炎イオン化検出器が開発され、ppmレベルのトレース分析が出来るようになり、環境や食品中の有害物質の探索に威力を発揮しています。特殊な原理ですが、PCBやダイオキシンなどハロゲン化合物には電子捕獲型検出器が考案され、ppb(10-9 v/v)の濃度が測定できる時代になりました。その外にもアルカリ熱イオン化検出器の名称で窒素やリンを含む成分を選択的に検出するものもあり、つぎつぎと開発が進んでいます。
3-2.熱伝導度検出器
有機元素分析では試料を熱分解して、生成ガスを定量することがほとんどですが、昔は吸収剤に成分を捉えてはかりに掛け、質量の増加を測定しました。質量は分銅によって標準化されますから、この方法は試料中の成分の絶対量を教えてくれます。しかしハンドワークを必要とするので、分析の自動化には向いていません。1960年頃からガスクロマトグラフが普及しはじめ、元素分析の主元素であるC、H、Nを水、二酸化炭素、窒素のクロマトグラムから定量しようとする研究が進められました(3-p7)。
もともとガスクロマトグラフィーは複数成分の分離のために開発された技術ですから、定性分析が主眼で,定量分析は付随したものでした。チャート紙上に描かれたピーク面積が成分量に対応することは容易に理解できましたが、その精密な測定法がすぐには実現されなかったことがその理由です。半値幅法、ピーク高さ法が定規と鉛筆で一応成分量に対応できるので、一般の化学研究室には普及しました。しかし有機元素分析のように究極の定量精度を要求するところでは、これらの方法は大まかでとても使い物になりません。ピーク面積を自動的に積算する装置も一方ではいろいろ試みられました。電子カウンターを使って刻々の信号強度を積算するのが一番精密には違いないのですが、4けたの積算数値を正確に打ち出す電子カウンターになると当時は随分高額のものになり、元素分析装置として普及できると思われませんでした。
もう一つの問題点は、当時標準的に使われた外経11mmの燃焼管の中で試料を燃焼させたとき、クロマトピークの最高点で検出器の信号が成分濃度に比例しているかどうかという事です。検出成分の種類にもよりますが、成分濃度は5% v/vあたりが上限といわれます。元素分析のような精密な定量分析には2~3%以下が安全と思われます。この制限のため,初期のガスクロマト方式の元素分析装置は試料量を1mg以下に設定していました(4-p325)。現在は超微量電子はかりがあって、さほど大きな障害ではありませんが、1960年代では厳しい壁と考えられました。
この頃自己積分法という新しい考え方が出て、上の二つの問題に解決策が現れました。まず燃焼ガスを一度一定体積の容器に入れ、検出器に適した濃度に均一にうすめ、このあとそれぞれの成分を検出器で持続的な信号として取り出す方法です。水、二酸化炭素、窒素は差動熱伝導度法という、煙道ガス中の特定成分のモニタリングに使われる技術を採用しました。この基本構想は1962年スイスのW.Simonらによって最初に提案されていますが(3-p9)、そのままではマニュアル操作が多く、自動化には向かないと思われていました。自動化を実現したのは1965年で、この年ペンシルバニア州立大学で国際微量化学技術シンポジウムがあり、ヤナコ分析工業とパーキンエルマが同時に公表しました。どちらも非公開で開発をしていたせいか、やり方は対照的で,前者がステンレス製のポンプを使って燃焼ガスを引き込んでいるのに対し、後者はガラス球に2気圧まで圧入するという方法をとっています。燃焼ガスの希釈体積が正確に再現しなければなりませんが、ポンプ法ではピストンのストローク距離で(4-p330)、ガラス球法では精密な圧力スイッチでこれを実現しました(4-p337)。
ポンプを使う方法は容器のピストンの気密保持が重要で、O-リングの材料やグリースの選択などに気をつけなければなりませんが、技術的には従来からある部品を使えばよいので、達成しやすいやり方です(3-p4)。初めはピストンの停止位置をマイクロスイッチで決めていましたが、後年光ビームの遮断による方法に代わり、一層ポンプ容積の再現性がよくなりました。ポンプ法のもう一つのメリットは装置の流路系をほぼ大気圧で操作できること、連結部などからのガス漏れの心配が少なく、かつ燃焼管入り口を常に開放できるので、試料導入に便利です(3-p4) 。
一方ガラス球法では機械的に動く部分がないので、装置構造が簡単になるメリットがありますが、2気圧近くまでキャリヤーガスを圧入し、正確にある圧力になった時ガスの注入を停止させなければなりません。圧力スイッチの精密なものが必要ですが、この部品はわが国では開発が遅れ、少なくとも1965年ごろには手に入るものではありませんでした。航空機や宇宙ロケットの先進国である米国では、電気系統の故障に備え空気配管によるニューマチック制御部品がいろいろ開発されていたようですから、これを利用しようという機運があったと思われます。
ポンプ法もガラス球法もそれぞれ手近にあって確実な方法を採用したのですから、定量分析の結果論としては優劣なしといえます。開発されて30数年どちらも基本的なやり方を変えずに現在に至っていますが、それだけ安心で頼りになる技術であった証拠でしょう。
クロマトグラフ法も自己積分法も熱伝導度検出器でヘリウムキャリヤー中の成分濃度を検知することに変わりはありません。ヘリウムは他の気体に比べて抜群に熱伝導度がよく、これに他の気体成分が混入すると熱伝導度が悪くなります。検出器は図11のようにステンレス鋼製のブロックにタングステンフィラメントをはめ込んだもので、フィラメントに電流を流すと熱を発し、この熱が周囲の気体を通って金属の壁に吸収されるとき、気体の熱伝導度によって熱の流れる量が変わります。ヘリウム中の成分濃度が高くなると、熱の伝達が悪くなるのでフィラメント温度が上がり、電気抵抗が同時に大きくなります。フィラメントを含むブリッジ回路を図12のように作っておくと電気抵抗の変化が不平衡シグナルとして検出されます(3-p53)。

図11 熱伝導度セル
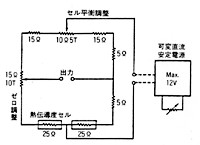
図12 ブリッジ回路(MT-2)
フィラメントにはMT-2型まで25Ωのタングステン線が用いられましたが、MT-3型以来60Ωになって感度が上がっています。またブリッジに流す電流によっても検出感度が大きく変わります。感度は電流の約2乗に比例すると言われますが、かといって感度を上げるためにむやみに電流を増やすと、フィラメントが変形したり極端なときは切断することもあります。精密な定量を継続的に続けるには200mAを超えないことが必要です。
ブリッジからの出力は目的成分が来ないときゼロか小さな値で安定していなくてはなりません。実際には燃焼管充填物からの僅かな物質の放出や連結部分からの空気の侵入などがあり、またブリッジ供給電流の変化、熱伝導度セルの温度漂動などでブリッジの出力はいつも一定というわけに行きません。ガスクロマト法ではベースラインドリフト、自己積分法ではゼロドリフトと呼んでいますが、ゼロ位置が次第に上昇したり下降したりすると、試料を燃焼したときに現れる成分シグナルの大きさの測定が困難になります。
3-3.ブリッジ出力の漂動
ブリッジ回路は正式にはホイートストンブリッジと呼びますが、図12のように電気抵抗を菱形に組み合わせ,上下方向に直流電流を流すと、左右のターミナルから僅かな電圧が発生します(図はMT-2型当時のもの)。始めにゼロ調整抵抗を動かして出力電圧をちょうどゼロとしておきます。この状態で回路の抵抗値のひとつが変化したとするとブリッジのバランスが崩れ、大きな出力になって現れます。
さて熱伝導度検出器のフィラメントをブリッジ回路の抵抗素子に組み入れると、ヘリウムキャリヤーのみを流しているときの出力を基準として、成分が乗ったときの信号出力が得られますが、基準を安定的に維持するためにいろいろな工夫がなされています。フィラメントは固定抵抗と違っていろいろな要因で抵抗値が変化します。一番大きな影響は検出器全体の温度変化です。ガスクロマトグラフ装置も初期から検出器の恒温槽が設けられ、制御精度は±0.1℃あたりから今は±0.01℃くらいになっていますが、CHN分析にはこれでもまだ足りないくらいです。
試料セルの温度だけを制御してもしきれないところは、同じ形のセルをブリッジの対照側に置き、試料セルの変化をキャンセルする方法を取っています。実際には試料セルと対照セルを同じステンレス製のブロックに並べて配置し、温度変化とフィラメントの抵抗変化を出力に出さないようにしています(図11)。
ガスクロマト方式では常に対照セルにキャリヤーガスのみを流し、試料セルに来る成分を含んだガスの熱伝導度を検出すればよいので,動作は比較的簡単ですが、成分はクロマトピークで与えられるので、その面積の測定という難問が控えています。かつては4桁の数値で面積を求めるためには高額の費用がかかりましたが、現在ではデジタルカウンタが進歩して5桁でもさほど費用はかかりません。むしろピークの先端の濃度で熱伝導度計の出力が直線性(比例関係)を失うことのほうが問題です。検量線を二次曲線で近似すればなんとか解決できますので、付属のコンピュータにそのプログラムを入れて分析結果を出すことができます。
CHNコーダーのような差動熱伝導度方式では、試料側に目的成分を含んだガスを通過させ、目的成分を除いたガスを参照側に導いて、その差を目的成分の濃度に対応させています。おおまかに考えると単純な原理で、事実煙道ガスのモニタリングには差動熱伝導度計の出力をそのまま成分濃度に置き換えて測定値としています。どうせ捨てるガスですから、迷惑がかからなければよいので、細かいことは考えていません。ところが有機元素分析に適用しようとすると、微妙なところで誤差が生じ、要求精度に一致しません。
CHNコーダーでは3対の差動熱伝導度計が設置されていて、流路の上流から水、二酸化炭素、窒素の検出が行われます。水検出器では試料側に3成分がありますが、水吸収剤で水を除去したガスが対照側に来ます。大気圧で測定していますから、水の無くなった分他の成分の濃度が僅かに高くなります。本来差動熱伝導法は目的成分を除去しても、その他の成分に関しては試料側と参照側に変化はないという原則で考えていますが、実際は除去されなかった成分の濃度が参照側でごく僅か高くなっているのです。二酸化炭素検出器や窒素検出器でも同様のことが起こっています(3-P55)(図13)。これらの現象ははっきりと数値的に解析が出来ますので、いくらの誤差が出るから、どう補正すればよいか理論計算が可能です。計算理論はかなり複雑な過程を経るので、ここでは省略しますが、「CHNコーダーの素顔」第6章;“補正と計算”(3-p51)に詳細記載されていますので、参照されることを期待します。ただし現実にはこの計算プログラムが装置付属のコンピュータに仕組んでありますので、何も考えなくても補正ずみの正しい分析結果を得ることができます。
複雑な補正計算はコンピュータがやってくれますが、オペレータにお願いすることが一つあります。それは試料側と対照側のセルが全く同じ感度で動作するよう調整することです。差動熱伝導度計は除去した成分だけに感じて、除去しない成分が試料側と参照側セルに同じ濃度で通過してもブリッジ出力とならない必要があります。もともと両方のセルにはマッチドペアというできるだけ直流抵抗の等しい二本のフィラメントを装填してあるのですが、実際電流を流すと両者が理想的に等しい感度で動作するとは限りません。そこでこの不均衡から生じるブリッジ出力を無くするため(図12)にはセルバランス抵抗が入れてあり、両者のフィラメント電流を加減して全く同じ感度で働くようにします(3- p53)。
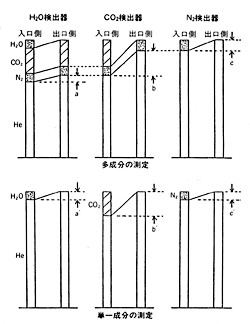
図13 差動熱伝導計による成分検出
セルバランスを取るには、おおざっぱにはセル電流を10mA前後変えてブリッジ出力の変化が最小となるところで固定してもよいのですが、厳密にはセル電流を設定値に固定し、乾燥空気2ml ほどを注射器などで燃焼管に注入すると、もしバランスが取れていなければブリッジ出力が変動します(ただし窒素検出器は空気注入の試験ができません)。少し面倒ですがブリッジ出力の変動が最小となるセルバランス抵抗の位置を見つけてください。新しい装置が導入されたときは最初の設定時にメーカーの方でバランスを取っておきますが、装置を実際に使いこむとフィラメントが汚れや酸化で特性の変化をしますので、精密な分析を常に期待するならば年に2~3回セルのバランスを取りなおすことが望まれます。
フィラメントは家庭の電球と同じで、消耗品と考えたほうがよいかも知れません。フィラメントが傷んでもセルのバランスを取れば一見うまく行っているように見えますが、これは風邪薬で熱を押さえながら仕事をやっているようなもので、少々危い話です。重厚なCHN分析計も,フィラメントが発する微妙な信号を頼りに分析データを得ているわけですから、できれば年に一度くらいはフィラメントの更新をしても十分値打ちがあるかと思います。更新時にはセルの内面や連結管の洗浄もやりますので、流路全体がこれですっきりきれいになるでしょう。
熱伝導度検出器はそこに成分ガスが来たことを知らせる部品ですが、来てもいないのに勝手に信号を出しては困ります。ベースシグナルの変動と一口にいわれますが、内容は複雑です。無視してもよい程度のものもありますが、対策の必要なものもあります。分析室の電源電圧が急に変わるとブリッジ電流にも影響がでる可能性があります。現在は直流電源に大容量のコンデンサーを入れて影響を押さえてありますが、これも限度があります。分析室の電源は十分余裕のあることと、電源経路に他の実験室の大型機械が含まれていないか注意すべきです。分析室の空調機が設置されているところでは、そこから吹き出す暖気や冷気の流れが直接元素分析装置に当たらないことが大切です。検出器の恒温槽は内部で高精度の調節を働かせているのですから、外部から温度を乱す要素を加えては調節能力が鈍ります。分析装置のない空間である程度平均化された雰囲気を作り、その雰囲気が静かに装置のほうに広がるような工夫をすべきです。
分析室の保安については事業所の方針でそれぞれ規制があり、夜間は電源を全部切るところや、小電力の通電を許すところがあります。検出器の恒温槽は出来れば夜間通電しておいたほうが、翌朝燃焼炉に電源を入れてからベースシグナルの安定が速くなります。しかしコンピュータに早朝起動の命令を入れておけば、出勤の1~2時間前にキャリヤーガスの供給を開始し、同時に電源の入力を自動的に行うことが出来ます。出勤時にはほぼベースシグナルは安定しており、すぐ捨て焼きなど管理作業が可能です。夜間の通電が規制されていてもこの程度の運用は許されると思いますので、活用されることをお勧めします。
4-1.ボルタンメトリー
聞きなれない術語かも知れませんが、ボルトメトリーとアンペロメトリーの合成語で、溶液に入れた二本の電極間の電位差を測るか、電極間に電流がいくら流れるかを測定する分析法です。ガラス電極pHメータは化学実験室の必需品になっていますが、これはガラス膜が水溶液中の水素イオンに感じて電位を高めるので、いつも一定電位を示す参照電極と組み合わせて一種の電池を構成し、その電位差を測っています。電位差はもし電極間に負荷をつなげば電流を流すことが出来ますので、起電力とも言われます。しかし本当に電流を流すと溶液中のイオン量も変化しますので、在るがままのイオン量を知るには、全く電流を流さない電位差計で電池の起電力を測ります。pHメータは従ってボルトメトリーの測定器ということができます(2-p138)(図14)。

図14 ガラス電極pH計
一方電極間に電流を流すアンペロメトリーでは溶液内でイオンの電気分解が行われます。銀イオン溶液中で電気分解すると、陰極で銀イオンは金属銀に還元され、陰極表面に析出します。この現象はファラデーの法則を生み出して古くから有名です。溶液中で1モルの銀イオンを全部金属銀にしてしまうと電気量は96486.7クーロンになり,この値は電気量の国際単位となっています。1アンペアの電流を1時間流すとその電気量は3600クーロンになります。電解電流と時間から電気量がわかりますが、その電気量から析出したイオンのモル数が出てきます。
話は簡単のようですが、実際には電気分解が進むと刻々目的イオンを溶液中から除いて行きますので、溶液内のイオン量は少なくなり、電解電流は減衰曲線を描きます(図15)。積算計で電解電流を測定しますが、どこまで減衰した時に積算を打ち切るかは最初に決めておかなければなりません。電気分解は目的イオンが電極で還元または酸化されることですが、複数イオンが存在すると目的外のイオンまで電解してしまいます。各イオンには決まった分解電圧というものがありますので、その電圧を2本の電極に加えなければなりません。ただ有機元素分析ではあまり複雑なイオン混合溶液を扱うことはないので、このあたりの知識は不必要かも知れません。

図15 電解電液の積算
電流を流すのでアンペロメトリーに属するのかも知れませんが、電気分解をしないので中途半端なものに電気伝導度の測定があります。最近イオンクロマトグラフィーが登場して脚光を浴びていますが、功績はイオン交換容量の極めて小さい樹脂を詰めたカラムにあるので、イオン濃度の測定に電気伝導度を使うことは昔からあった古い技術です。ここでは電極間に1キロヘルツ前後の交流をかけ、陽イオンと陰イオンが電極の間を行ったり来たりして電極の電荷を運んでいます。直流ですと陽極と陰極で電気分解などが起こりますが、交流では高速に極性が反転するので電気分解を起こす暇がありません。一定電圧をかけたとき交流電流の大きさが溶液内のイオン濃度に対応しますので、イオンクロマトグラムの図形が得られます。
この外にも電気分析のレパートリは多岐にわたります。1900年代の始めチェコスロバキアの J. Heyrovsky によって発明されたポーラログラフィーは、水銀滴下電極を用い、水銀球が毛細管の下端で成長し落下するまでの間に周囲のイオンを電解し、電解電流を連続的に記録しました。水銀電極の電位をゆっくり変えて行くと、電解されやすいイオンから順に階段状にポーラログラムが得られ、その図形の解析から複数イオンの定性と定量が出来ました(2- p155)(図16)。当時としては画期的な大発明で、今日の機器分析のさきがけと言ってよいでしょう。1959年その功績によってノーベル化学賞を受賞しています。残念ながらその後原子吸光やプラズマ発光など新しい分析法に切りかわり、最近はポーラログラフィーを利用する機会が随分減っています。
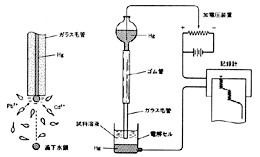
図16 ポーラログラフ装置の基本構成
もう一つアノーディックストリッピングボルタンメトリーという長い名前の手法が高感度電気分析法にあります。詳しい説明は省きますが、要するに超希薄な金属イオン溶液を白金電極で長い時間電解し、陰極に金属を集めておいて、このあと電極の極性を逆にすると陰極が陽極になり、貯めてあった金属が一挙にイオンになって溶け出します。ゆっくり貯めたものを一挙に吐き出すのですから、極めて高感度の測定が可能です。環境、排水、食品などに含まれる有害物質のモニタリングにはよく使われている方法です。
4-2.電位差測定
乾電池は新しいもので1.5ボルトの起電力がありますが、中心の炭素棒(陽極)と容器の亜鉛(陰極)の間に1.5ボルトの電位の差があることを示しています。つまり電池とは異なる電位を持つ2本の電極を組み合わせたものです。それでは電極電位とは何かとなると説明がかなり面倒になりますが、簡単にいえば電極にどのくらい電子が余分にたまっているかの目安と考えて差し支えないでしょう。例えば亜鉛の電極を塩化アンモニウムなど電解質溶液に漬けると金属亜鉛が亜鉛陽イオンとなって少し溶け出します。この時電子を電極に残して出て行きますので、亜鉛電極中に電子が余分にたまります。こうなると逆に電極は溶液中に出てゆこうとする亜鉛イオンを引っ張り戻す力が働き、これ以上亜鉛は溶液に溶け出しません(図17)。新しい乾電池の亜鉛電極はこの状態にあります(2-p125)。

図17 亜鉛電極の反応
乾電池を電球など外部回路につなぎますと、電極内の電子が流れ出てしまいますから、亜鉛電極は亜鉛イオンを引き止める力が無くなり、どんどん溶液の方に溶け出します。外部回路を切ると再び電極に電子がたまり亜鉛イオンの溶出は止まります。しかし溶液内の亜鉛イオンが増えていますから、電極から亜鉛イオンになって溶液に溶け出す力が減り、亜鉛電極には前ほど電子がたまらず、電極電位はプラスの方向に移ってきます。
炭素電極も似たようなことをしますが、電気化学反応が少し複雑なのでここでは省きます。しかし古くなると陽極付近の反応物質が減少して電極電位はマイナスの方向に移り、結果として亜鉛電極との電位差が小さくなって0.9ボルトになるなどという原因を作っています。ここで気がつくことは、われわれは電池の起電力を測って能力を評価することができますが、起電力を構成する2本の電極の電位は単独には測定できないということです。A-B=X の式で、X を知っても A、Bの値は不明と言うのと同じです。
こういうとき人間は標準を作って対応します。標準水素電極がそれで、その電極電位はいつもゼロボルトと取り決めます。Bがゼロですから、これと組み合わせる任意の電極の電位は起電力Xと同じで、この方法で電極電位が決められます。標準水素電極は1モル塩酸に浸漬した白金電極に水素ガスを吹き込みながら作るもので、物理化学的には基準になりえますが、実用上はいつも水素ガスが要るので不便です。
そこで登場するのが取り扱いやすい2次標準で、銀―塩化銀電極が広く使われています(1- p243)。その電極電位は25℃において+0.222ボルトです。いつも変わらずこの電位を示すので、参照電極と呼ばれています。pH測定でも例外ではありません。ここではガラス電極と銀―塩化銀電極を組み合わせ、その起電力を測っています。ガラス表面が試料溶液中の水素イオン濃度に感応して電位を変化させるので、参照電極の電位との差を測って酸―塩基の度合いを知ることができます(5-B378)(図18)。
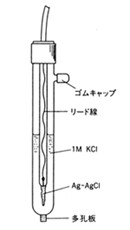
図18 銀-塩化銀電極
感応電極の種類を変えるといろいろなイオン種の測定ができます。水素イオン以外によく測定されるのは銀イオン、塩素イオン、フッ素イオン、ナトリウムイオン、カリウムイオンなどです。電極とイオンとの電気化学反応はそれぞれ違っていて、ここで全部を解説する余裕はありませんが、デリケートな電極の表面反応ばかりですから、表面の手入れには特に留意の必要があります。
pHガラス電極は常に水に浸けておかなければなりません。ビーカーの水に浸けたときは、水の補給を忘れないことが肝心です。また長期しまい込むときは水を入れたゴムキャップで感応部分を覆っておきます.銀電極は表面に酸化膜やハロゲン銀膜が付きやすいので、使用に先立って細かいサンドペーパーで金属光沢が出るまでこすります。塩素イオンやフッ素イオン電極は円盤状の感応素子が先端にはめ込まれていますが、表面を鹿皮で丁寧にこすり、よごれなどを落とします。ナトリウムイオン電極は一種のガラス電極で、平素は0.1モル程度の食塩水に浸けておきますが、カリウムイオン電極は全く構造の違う油性の液膜電極で、乾燥状態でも保存できます。感応部分がカートリッジになっていますので、動作が不良になったときはその交換ということになります。ただし使用の前には2時間ほど0.1モル程度の塩化カリウム溶液に浸漬しておきます。
感応電極は測定の主役ですから、誰でも大切に扱うことは当然ですが、起電力は参照電極との電位差であることを忘れてはなりません。参照電極が絶対不変の電位を守ってくれればよいのですが、そうも行かないのが現実です。こちらの方の面倒も見なければ、起電力を安心して感応電極の電位とすることが出来ません。
一番よく起こるトラブルは複合型ガラス電極(5-B335)(図19)によく使われるピンホール型液絡の参照電極で、液絡部分が乾くと内部の塩化カリウムが滲み出し、結晶がピンホールに詰まって液絡の電気伝導性を失わせます。切れてしまえば起電力そのものも不安定になって測定中に気が付きますが、そこへ行くまでは案外気が付かず、感応電極のほうが悪いように思ったりします。水に漬けておくと結晶が溶けて回復することもありますが、孔の中で固まってしまって元に戻らないこともあります。このときは新しいものと取り替えるしかありません。
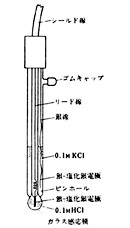
図19 複合型ガラス電極
(図18)のようにセラミックのフィルター材料を液絡に使ったものは、ピンホール型より詰まりの現象が少なくてよいのですが、内部の塩化カリウムの滲み出しがやや多いので、試料溶液を汚染します。それで目的物の測定上差し支えがなければよいのですが、一応検討しておかなければなりません。もう一つスリーブ型というのもあります。これは毎日使う前にすり合わせ部を緩め、内部液を適当に流し液絡を作りますので確実ですが、うっかり使わずに放置するとすり合わせに塩化カリウム結晶が固着し、このあとどうにも緩まないという事故があります。使わないときも水に浸けておくことと、時々すり合わせを緩めて見ることが必要です。塩素イオンの測定のときはダブルジャンクション参照電極(5-B340)(図20)を用いたほうが安全です。このときは外側の内部液に硝酸アンモニウムなどを入れておきます。
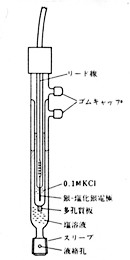
図20 ダブルジャンクション参照電極
4-3.電気伝導度測定
すでに述べたように、溶液の電気伝導度の測定は電気化学反応を伴わないので、電気化学分析の項目に入れるのはおかしいかも知れません。しかし有機元素分析の分野でイオンクロマトグラフィーがハロゲン、硫黄、アルカリ金属の分析に一部使われるようになって、その検出器に電気伝導度計を使うことから無関心ではいられなくなりました。イオンクロマトグラフィーは原理的にはイオン交換クロマトグラフィーなのですが、イオン交換基が充填粒子の表面だけに作ってあって、内部に入り込まないため、保持時間が短く、鋭い分離のピークが得られます。
成分の検出には吸光度を測るものもありますが、無機イオンには電気伝導度計が一般的に用いられます(6-p162)。希薄な目的イオンを測定するので、検出器を通る溶離液は電気を伝えないことが必要で、このため巧妙な工夫がしてあります。サプレッサーといわれる部品がそのために使われます。たとえば炭酸水素ナトリウムを溶離液とすると、薄い半透膜を介して硫酸液と接したとき、硫酸イオンは半透膜を通過できませんが、ナトリウムイオンは硫酸のほうに抜けて行き、代わりに水素イオンが入ってきます。こうしてできた炭酸イオンは結局水と二酸化炭素になりますから、電気伝導性もなくなります。
NaHCO3 + H2SO4 → H2CO3 + NaHSO4
H2CO3 → H2O + CO2
サプレッサーにはネフィオンという高分子膜が使われますが、はじめは中空糸を用いたものの、ピークの広がりがあるのでまもなく平板状のものに切りかわりました(図21)。サプレッサーはかなり高度の機能を持つ部品ですから、これを省略するため溶離液に酒石酸ナトリウムなど電気伝導度の低いものを用い、サプレッサーなしでイオンクロマトを行う装置も市場にあります。ノンサプレッサー方式とも呼ばれていますが、元素分析にはやはりサプレッサー方式を用いたほうが安全です。ハロゲンの定性、定量ができるので、フラスコ燃焼のあと吸収液の分析に活用されています。カラムを変えると陽イオンの分析もできるので、将来性があります。

図21 薄膜サプレッサー
電気伝導度は溶液中のイオンの移動速度(易動度)に依存しています。易動度はイオンの種類によっても違いますが、温度によってもかなり変化します。もし定量を元素分析のように高い精度で行おうとするならば、検出器の温度制御をこれに見合うものとしなければなりません。皮相的な見方ですが,安価な装置は温度制御もそれだけ気軽に作ってあります。
検出器自体は完全なハードウエアで中を見ることはできません。イオンクロマト装置では通常の電気伝導度計のセルと違って、デッドボリュウムの極端に小さいものを使用しています。セルの内面や電極の汚れも気になりますが、もし問題が起きたとしても手を入れることはできませんので、メーカーの技術者に点検,修理を委託するしかありません。
5-1.おわりに
20世紀は人間の感覚器をセンサーに置き換えた時代ということもできます。日が暮れると門灯が点くといった単純なセンサーから、微弱な光の強度や希薄な気体の濃度を1/1000の精度で測り出すなど、人間の感覚器の及びもつかない機能を持ったものまで作り出されました。しかし高度のセンサーほど外からの妨害や自身の不安定性がクローズアップされてきます。それにもかかわらずセンサーの出力から貴重なデータを得て、われわれは日々の職能を果たしているのですから、センサーの機能の維持,管理について絶えず知識を蓄え、努力を続けるべきです。
6-1.参考文献
1)穂積啓一郎,北村桂介:機器分析通論,広川書店 (1980).
2)穂積啓一郎,吉村菊子:機器分析のとびら,さんえい出版 (1995).
3)ヤナコ分析工業:CHNコーダーの素顔,さんえい出版 (1993).
4)日本分析化学会有機微量分析研究懇談会:有機微量定量分析,南江堂 (1969).
5)日本公定書協会:第13改正日本薬局方解説書,広川書店 (1996).
6)花岡 譲:ぶんせき, (1986),162.